Chapter Outline
DEFINITION AND CLASSIFICATION OF PHAGOCYTES
PHAGOCYTE DISTRIBUTION AND STRUCTURE
Humoral Mediators of the Inflammatory Response
Adhesion and Migration into Tissues
Recognition, Opsonization, and Phagocytosis
Cytocidal and Digestive Activity
Specialized Functions of Mononuclear Phagocytes
Specialized Functions of Eosinophils and Basophils
Pathologic Consequences of Phagocyte Activation and Inflammatory Response
QUANTITATIVE GRANULOCYTE AND MONONUCLEAR PHAGOCYTE DISORDERS
DISORDERS OF GRANULOCYTE AND MONONUCLEAR PHAGOCYTE FUNCTION
Disorders of Opsonization and Ingestion
Disorders of Neutrophil Granules
Disorders of Oxidative Metabolism
Disorders of Cytokines and Impaired Phagocyte Function
Definition and Classification of Phagocytes
Phagocytic leukocytes are bone marrow-derived cells that have the capacity to engulf and digest particulate matter. Phagocytes are essential for the host response to infection and injury and are equipped with specialized machinery enabling them to seek out, ingest, and kill microorganisms. Other functions include the synthesis and secretion of cytokines, pyrogens, and other inflammatory mediators, as well as the digestion of senescent cells and debris. These functions are important for resolution of injury and wound repair as well as linking innate to adaptive immunity.
The phagocyte system has two principal limbs: granulocytes (neutrophils, eosinophils, and basophils) and mononuclear phagocytes (monocytes and tissue macrophages). Both limbs participate in innate immunity and initiation of acquired immune responses. Neutrophils, the rapid effector cells of the innate immune system, circulate in the blood stream until encountering specific chemotactic signals that promote adhesion to the vascular endothelium, diapedesis into tissues, and migration to sites of microbial invasion or tissue injury. Mononuclear phagocytes also function as resident cells in certain tissues, such as lung, liver, spleen, and peritoneum, where they perform a surveillance role. This chapter is divided into three major sections. The first describes the normal distribution, structure, and function of granulocytes and mononuclear phagocytes. The second section reviews the clinical disorders associated with deficient or excessive phagocytic number. The third section focuses on disorders of phagocyte function, including both intrinsic phagocyte defects and conditions secondary to other disease processes.
Phagocyte Distribution and Structure
Regulation of Myelopoiesis
Granulocytes and monocytes are produced in the bone marrow in a complex, highly regulated, and dynamic process that requires both specific hematopoietic growth factors and an appropriate bone marrow microenvironment. As reviewed in Chapter 6 , multipotent, self-renewing hematopoietic stem cells (HSCs) give rise to lineage-restricted progenitor cells that divide and further differentiate in the bone marrow before their release into the intravascular compartment. Transcription factors of the PU.1 and CCAAT/enhancer binding protein (C/EBP) families play prominent roles in normal myelopoiesis ( Fig. 22-1 ). PU.1 is important for the development of early myeloid precursors and is absolutely essential for subsequent differentiation of the monocyte/macrophage lineage. Early steps in the differentiation of granulocytes are dependent upon C/EBPα, whereas C/EBPε activity is required for terminal maturation beyond the metamyelocyte stage. Cytokines that promote the proliferation and differentiation of neutrophils and monocytes from primitive precursor cells include interleukin (IL)-3, IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage CSF (M-CSF), and granulocyte CSF (G-CSF). The latter two cytokines are relatively specific for the monocyte and neutrophil lineages, respectively. When apoptotic neutrophils are taken up by macrophages in tissues, they deliver a negative feed back via T-helper type (Th17) lymphocytes that results in reduction of G-CSF production. During infections, activated macrophages release cytokines such as IL-1, IL-6, and tumor necrosis factor (TNF) that activate stromal cells and T lymphocytes to produce additional amounts of CSFs and increase the production of myeloid cells. This is termed emergency granulopoiesis and in mice has been shown to depend on G-CSF activation of Stat3 and on C/EBPβ expression in granulocyte monocyte precursors (GMPs). However, in vivo feedback is impaired in Tlr4−/− and Trif−/− , but not MyD88−/− animals, thereby directly linking TLR-triggering to granulopoiesis. IL-5 and IL-3 are the principal cytokines inducing human basophil growth and differentiation. In addition to their regulatory role in hematopoiesis, hematopoietic growth factors can act on mature myeloid cells and stimulate their functional activities and survival.
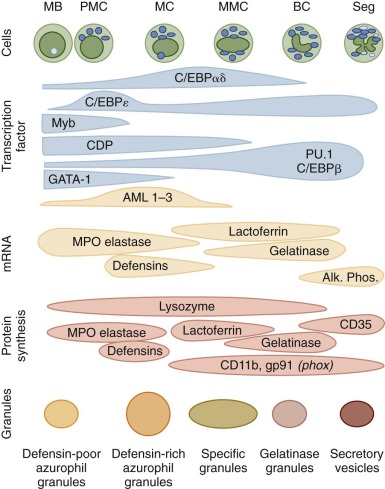
Myeloid differentiate also appears to be modulated by retinoic acid receptors and by transcriptional repressors such as Gfi-1. The participation of retinoic acid in myeloid development was originally surmised from its ability to induce differentiation of myeloid leukemia cell lines and leukemic promyelocytes in patients with acute promyelocytic leukemia, as discussed in Chapter 11 .
Micro–ribonucleic acids (miRs) such as miR21, miR29a, miR125, miR130a, miR146, miR155, miR196b, and miR223 participate at several levels of myelopoiesis and in control of lineage decisions in the bone marrow. MiRs are expressed at different stages during myelopoiesis, and several clusters can be identified depending on their expression profile during granulopoiesis. Gfi-1 is a key repressor of miR21 and miR196b expression. Expression of these miRs promote monocytic differentiation and block granulocytic differentiation. In addition to the well-known repression of translation, miRs can also regulate transcription by controlling the accessibility of promoters by epigenetic modifications as demonstrated on the nuclear factor IA promoter, which is silenced by miR223, allowing for terminal granulocytic differentiation.
Granulocytes
Neutrophils
The neutrophil life span is traditionally divided into the bone marrow, circulating, and tissue phases. Approximately 14 days are spent in the bone marrow, where proliferation and the early stages of neutrophil differentiation are followed by the final stages of maturation and retention in a large, nonmitotic storage pool that is many times larger than the circulating and tissue neutrophil populations ( Table 22-1 ). Release is regulated by chemokines expressed on the cells, and their ligands are expressed by stroma cells. CXCR4 and its ligand CXCL12 retain cells, and mutations in the CXCR4 receptor account for the warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome, an inherited neutropenia. Whereas CXCR2 and its ligands CXCL1 and CXCL2 promote neutrophil release. Also, the integrin α 4 β 1 , known as very late antigen 4 (VLA-4) may tether bone marrow neutrophils to vascular cell adhesion molecule 1 (VCAM-1) expressed on bone marrow stromal cells. Once released into the bloodstream, neutrophils have an estimated half-life of 6 to 10 hours and move between circulating and marginated pools in a reversible fashion. These estimates are based on several independent determinations using both in-vivo and in-vitro labeling techniques and transfusions, and they agree well with the estimates of neutrophils production rates of 10 cells/kg/day and circulating neutrophil counts 2.5 to 4 × 10 9 /L. A recent controversial report suggests a half-life of 4 to 5 days both in humans and mice. Neutrophils exit circulation by diapedesis between or through endothelial cells into tissue sites of infection or inflammation. Once in the tissues, neutrophils are believed to live for another 1 to 2 days before undergoing apoptosis and engulfment by macrophages or they form neutrophil extracellular traps (NETs) consisting of deoxyribonucleic acid (DNA) and antibiotic proteins from nucleus, granules, and cytosol in a regulated process called netosis that traps microorganisms. This is discussed in more detail later in this chapter.
| Transit Time Range (hr) | Total Cells (×10 9 /kg) | |
|---|---|---|
| Neutrophils | ||
| Marrow Mitotic Compartment | ||
| Myeloblast | 23 | 0.14 |
| Promyelocyte | 26-78 | 0.51 |
| Myelocyte | 17-126 | 1.95 |
| Postmitotic Marrow Maturation and Storage Compartment | ||
| Metamyelocyte | 8-108 | 2.7 |
| Band | 12-96 | 3.6 |
| Neutrophil | 0-120 | 2.5 |
| Total storage | 8.8 | |
| Vascular Compartment | ||
| Circulating neutrophils | 4-10 | 0.3 |
| Marginated neutrophils | 4-10 | 0.4 |
| Total blood neutrophils | 0.7 | |
| Tissue compartments | 0-3 days (?) | Not known |
| Neutrophil turnover rate | 1.6 × 10 9 /kg/day | |
| Monocytes | ||
| Marrow mitotic compartment: promonocyte | ≈160 | 0.006 |
| Postmitotic marrow compartment: monocyte | 24 | 0.10 |
| Vascular compartment | 36-104 | 0.024 |
| Tissue compartment | Days-months | Not known |
| Monocyte turnover rate | 1 × 10 8 /kg/day | |
Myeloblasts are the earliest morphologically recognizable granulocyte precursors in the marrow and are identified by their relatively undifferentiated appearance with a large, oval nucleus, several prominent nucleoli, and few or no granules in a gray-blue cytoplasm in Wright-stained preparations. This stage of neutrophil differentiation is followed by the promyelocyte and myelocyte stages, which are distinguished by the appearance of distinct neutrophil granule populations ( Table 22-2 ). Azurophilic, or primary, granules are formed during the promyelocyte stage and contain myeloperoxidase (MPO), bactericidal peptides, and lysosomal enzymes. The subsequent myelocyte stage is distinguished by the formation of peroxidase-negative specific, or secondary, granules containing lactoferrin. No further cell divisions occur after the myelocyte stage. The metamyelocyte, band, and mature neutrophil exhibit progressive nuclear condensation, accumulation of glycogen, and accumulation of tertiary, gelatinase-rich granules and secretory vesicles that are endocytic vesicles marked by albumin and Complement Receptor 1 (CR1) (CD35). These neutrophil precursors can be identified and isolated by flow cytometry based on their surface antigen profile and forward and side scatter.
| Primary: Azurophil Granules | Secondary: Specific Granules | Tertiary: Gelatinase Granules | Secretory Vesicles |
|---|---|---|---|
| Membrane | |||
|
|
|
|
| Matrix | |||
|
|
| Plasma proteins |
In Wright-stained blood smears, the mature neutrophil is 10 to 15 mm in size with a multilobed, polymorphic nucleus that has highly condensed chromatin and a yellow-pink cytoplasm containing numerous granules as well as clumps of glycogen. The mean lobe count is usually slightly less than three. Circulating neutrophils appear round with some cytoplasmic projections and surface ruffling. The morphologic changes seen with neutrophil differentiation are accompanied by temporally coordinated changes in gene expression and protein synthesis (see Fig. 22-1 ). Transcription and translation of messenger RNAs (mRNAs) for MPO and cathepsin G, which are both primary granule constituents, are restricted to myeloblasts and promyelocytes. In contrast, expression of the secondary granule proteins such as lactoferrin and transcobalamin I occurs in myelocytes and metamyelocytes. Gelatinase expression occurs even later in maturation and is first detected in bands and bone marrow neutrophils. The leukocyte β-integrin subunit CD11b is first detectable in myelocytes, and increases throughout the later stages of neutrophil differentiation. The gp91 phox subunit of the respiratory burst oxidase complex is expressed relatively late in neutrophil maturation, consistent with the observation that respiratory burst activity is not detected until the metamyelocyte stage.
The mature neutrophil, previously thought of as an “end-stage” cell, retains the capacity for inducible gene expression and protein synthesis even after release from the marrow cavity. Diapedesis and exposure to cytokines induce neutrophil expression of mRNA transcripts for IL-1, IL-6, TNF-α, GM-CSF, M-CSF, and IL-8, which may promote recruitment and activation of both phagocyte and lymphocyte populations in the inflammatory response.
Abnormalities in Neutrophil Morphology.
Upon neutrophil activation by inflammatory signals, granule fusion can result in vacuolization and toxic granulation (prominent azurophilic granules). These morphological changes reflect a nonspecific response to inflammation and do not necessarily indicate the presence of bacterial infection. Large azurophilic granules but normal specific granules are seen in the Chédiak-Higashi and the Alder-Reilly anomalies (both autosomal recessive [AR] traits), but neutrophil function does not appear to be affected in the latter. Döhle bodies can be seen in normal neutrophils at times of infection. These inclusions represent strands of rough endoplasmic reticulum that are retained from a more immature stage and stain bluish because of their high content of RNA and ribosomes. Döhle bodies in granulocytes and monocytes, in combination with leukopenia, giant platelets, and variable thrombocytopenia, characterize the May-Hegglin anomaly. This autosomal-dominant syndrome, like the similar Fechtner and Sebastian syndromes, is caused by mutations in the gene encoding non–muscle myosin heavy chain 9.
Neutrophil hypersegmentation can be a sign of vitamin B 12 or folate deficiency. Hypersegmentation is also reported in small subset of neutrophils in the circulation that have reduced expression of L-selectin (CD62L). These may represent a more mature subset approaching senescence and also are reported to be capable of suppressing T-cell activation during inflammation. Hypersegmented neutrophils with a mean of four lobes also occur as a rare autosomal dominant (AD) trait that is not associated with disease. Nuclear hyposegmentation is seen in Pelger-Huët anomaly, an AD trait caused by mutations in the gene encoding the lamin B receptor, an integral protein of the nuclear envelope. Typically the nucleus is bilobed (often described as pince-nez ) but has mature, coarse, and densely clumped chromatin. The nucleus remains round in the rare homozygote. Pelger-Huët anomaly must be distinguished from neutrophil band forms and from the acquired or “pseudo” Pelger-Huët form that can be seen with myeloproliferative disorders. Bilobed neutrophil nuclei are also seen in a rare functional disorder of neutrophil maturation, specific granule deficiency (SGD). In this disorder, the pink-staining specific granules are absent in peripheral blood neutrophils. Giant granules representing defective membrane targeting of proteins in secretory lysosomes are seen in Chédiak-Higashi syndrome (CHS) neutrophils, most prominently in the bone marrow. SGD and CHS are discussed in more detail later in this chapter.
Neutrophil Granule Biosynthesis and Classification.
The numerous intracellular granules and vesicles in the neutrophil cytoplasm function as storage pools for cell surface receptors and as reservoirs of sequestered digestive and microbicidal proteins. Many compounds are multifunctional. For example, cathepsin G defensins and azurocidin are both antimicrobial and chemotactic for monocytes and T cells, which helps amplify the inflammatory response and link innate to adaptive immunity. The older classification of granules as either peroxidase-positive (azurophilic or primary) and peroxidase-negative (specific or secondary) has proven to be too simplistic. Instead, neutrophil granules are a continuum from the earliest appearing azurophil granules formed in immature promyelocytes to gelatinase granules formed in band cells. The content of granules reflects the transcriptional profile during terminal neutrophil differentiation in the marrow. A current classification of neutrophil granules is shown in Table 22-2 , which summarizes the composition of their membranes and luminal (matrix) contents.
Azurophilic (primary) granules are defined histochemically by the presence of MPO, an enzyme in the oxygen-dependent killing pathway. This green heme enzyme lends it color to collections of mature neutrophils (pus) or myeloid leukemia cells in the bone marrow or extramedullary tumors (“chloromas”). Azurophilic granules also contain peptides and proteins that participate in oxygen-independent killing of microbes. Other components of the azurophilic granule matrix include neutral serine proteases and other digestive enzymes characteristic of lysosomes.
Specific (secondary) granules, which are uniquely found in neutrophils, are classically identified by their content of lactoferrin, an iron-binding protein that also has direct bactericidal activity. Specific granules also contain additional antibiotic substances, including lysozyme, lipocalin 2 (also known as neutrophil gelatinase–associated lipocalin; NGAL), a bacterial siderophore-binding protein, and the metalloproteases collagenase and gelatinase. The membrane of the secondary granules contains a major proportion of the neutrophil’s supply of flavocytochrome b558 , the electron carrier of the respiratory burst oxidase. Specific granule membranes also contain a pool of receptors for adhesive proteins, TNF, and chemotactic formyl peptides.
Although specific granules contain collagenase and some gelatinase, most of the neutrophil’s store of gelatinase is localized to the matrix of gelatinase (tertiary) granules, which also contain the membrane-associated metalloproteinase leukolysin (MMP-25). Tertiary granules are formed relatively late in neutrophil differentiation and are smaller and more easily mobilized for exocytosis than secondary granules. Secretory vesicles are formed in bands and mature neutrophils by endocytosis of the plasma membrane and serve as an important store of leukolysin as well as the adhesive protein Mac 1 (CD11b/CD18) and many other membrane receptors (see Table 22-2 ). Proteomics has permitted a more global view on neutrophil granule proteins.
Neutrophil Cell Surface Receptors.
A primary function of the mature neutrophil is to move rapidly into tissue sites to destroy invading microbes and clear inflammatory debris. To respond to inflammatory stimuli, the neutrophil is equipped with an array of cell surface receptors for adhesive ligands, chemoattractants, and cytokines that can be divided into groups based on their structure and the major intracellular signaling pathway to which they are linked ( Table 22-3 ). Many of these surface proteins are pattern-recognition molecules such as Toll-like receptors (TLRs) and formyl peptide receptors, reflecting the neutrophil’s role in the innate immune response. Microdomains in the plasma membrane also known as lipid rafts, which are enriched in cholesterol, glycosphingolipids and glycosylphosphatidylinositol (GPI)-anchored proteins, and CD11b/CD18, can function as pattern recognition structures by virtue of their glycosphingolipids such as lactosylceramide (CDw17), the lipopolysaccharide (LPS)-binding GPI-anchored protein CD14 and the carbohydrate-binding domain of CD11b. These aggregates can then signal intracellularly via the associated integrins but likely also via lipid tail interactions of CDw17 and the tyrosine kinase Lyn attached to the inside of the membrane via its lipid-tail.
| Receptor Grouping | Examples | Structural Characteristics |
|---|---|---|
| G-protein linked | fMLP, C5a, PAF, LTB 4 , IL-8, chemokines | Seven-transmembrane–spanning domains (serpentine); linked to heterotrimeric GTP-binding proteins |
| Membrane tyrosine kinases | PDGF | Integral membrane protein, intrinsic tyrosine kinase activity; ligation leads to receptor dimerization and cross (“auto”) phosphorylation |
| Tyrosine kinase linked | FcγRIIa, GM-CSF | FcγRII is a member of the immunoglobulin family of receptors The GM-CSF receptor is an 84-kD transmembrane protein related to receptors for IL-2 and IL-6 Ligation of receptor activates cytosolic tyrosine kinases |
| GPI linked | FcγRIIIb, DAF, CD14 | Receptors with no transmembrane or intracellular domains. May associate with a partner receptor to mediate signal transduction |
| Adhesion molecules | β 2 Integrins L-selectin | β Integrins are heterodimers with relatively long cytoplasmic tails L-selectin has an extracellular lectin-binding domain and a very short cytoplasmic tail Ligation results in potentiation of the oxidative burst and phagocytosis in adherent cells, calcium signaling, actin cytoskeletal changes, and upregulation of gene expression |
| Ceramide linked | TNF | Single-membrane-spanning glycoproteins; ligation activates membrane-bound sphingomyelinase with generation of ceramide, which in turn activates a 96-kD protein kinase |
The signal transduction cascades triggered upon ligand binding to neutrophil receptors are complex and probably redundant. TLRs recognize structures specific for microorganisms, the pathogen-associated molecular patterns (PAMPs) such as LPS, lipoteichoic acid, and flagellin, but they also host molecules associated with cellular stress and injury, the damage-associated molecular patterns (DAMPs) such as heat-shock protein 60 and DNA. A common early event downstream of neutrophil receptor binding is activation of phospholipase C (PLC), which hydrolyzes the membrane phospholipid, phosphatidylinositol 4,5-bisphosphate (PIP 2 ) to generate two important second messengers, diacylglycerol and inositol 1,4,5-triphosphate (IP 3 ), which in turn cause release of calcium from intracellular stores and activate protein kinase C. Changes in intracellular calcium concentration are important for neutrophil degranulation and secretion and for phagolysosome fusion during phagocytosis. Activation of phosphoinositide 3′-kinase (PI3K) is another common early event that catalyzes the phosphorylation of PIP 2 to generate a third important lipid messenger, phosphatidylinositol 3,4,5-trisphosphate (PIP 3 ). Neutrophil activation is also accompanied by alterations in the phosphorylation status of intracellular proteins, as regulated by protein kinase C, tyrosine kinases and phosphatases, and serine/threonine kinases of the mitogen-activated protein (MAP) kinase family. Guanine nucleotide binding proteins play important roles in neutrophil signal transduction. These include the heterotrimeric guanosine triphosphate (GTP)-binding proteins that are coupled to the seven transmembrane-spanning domain (serpentine or heptahelical) receptors for chemokines and other chemoattractants and the low molecular weight guanosine triphosphatases (GTPases) of the Ras superfamily. The latter category includes p21Ras itself, which can be activated via chemoattractant receptors, and the Rho family GTPases Rho, Rac, and Cdc42, which are involved in the regulation of many neutrophil responses, including adhesion, the respiratory burst–reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and actin remodeling during migration and phagocytosis. A dominant-negative form of the Rac2 GTPase has been identified in an infant with recurrent deep-seated bacterial infections and leads to multiple defects in phagocyte function.
Signaling through these receptors is subject to positive and negative modulation. Triggering receptors expressed on myeloid cells (TREMs), of which TREM-1 is expressed on neutrophils, potentiate the signaling through TLRs. TREMs signal through an intracellular adaptor protein, DNAx activating protein (DAP) of 12 kD, DAP12. DAPs contain an immunoreceptor tyrosine–based activation motif (ITAM), which when phosphorylated, actives the protein tyrosine kinase Syk, which activates PI3K and phospholipase Cγ (PLCγ) via Bruton tyrosine kinase and thus potentiates the signals generated through TLRs, chemokine receptors, and integrins.
Neutrophils express high levels of G-CSF and GM-CSF receptors. These receptors signal through Janus kinases (JAKs), which dock to phosphorylated tyrosine residues in the cytoplasmic tail of the receptors, and from there phosphorylate and activate STAT-3 and -5 that induce transcription of several genes, in particular cytokines. The signaling through receptors activated via tyrosine phosphorylation is modulated by two different mechanisms, suppressors of cytokine signaling (SOCS) and immunoreceptor tyrosine–based inhibitory motifs (ITIMs). SOCS-3 is induced by activated STAT-3 and binds to and blocks phosphotyrosines in the activated growth factor receptor. In contrast, activation of receptors such as signal inhibitory receptor on leukocytes-1 and CD300a that contain an ITIM recruits phosphotyrosine phosphatases to dephosphorylate the activated cytokine receptors and reduce signaling.
Neutrophil Subsets.
Circulating neutrophils are traditionally viewed as one homogenous population. However, it is known that two subsets can be identified based on the presence or absence of CD177. The fraction of neutrophils that express this antigen varies from 0 to 100 amongst individuals but is stable through life. CD177 is the surface receptor for proteinase 3, the antigen associated with Wegener granulomatosis, and signals generated via CD177 are believed to play a major role in this disease. As mentioned, a subpopulation of hypersegmented neutrophils that also have reduced L-selectin expression has been reported, which may indicate a more mature neutrophil population with T-cell suppressive activity. Recently, a specific granule protein with antibacterial properties, OLFM4 was found to be expressed in 25% of human granulocytes with wide but constant variation among individuals.
Eosinophils
Like the neutrophil, the eosinophil is compartmentalized in the bone marrow into mitotic and storage pools; these usually constitute no more than 0.3% of the nucleated bone marrow cells. Eosinophils arise from a progenitor cell, the CFC-Eo, that is committed at a relatively early stage to differentiate into eosinophils instead of neutrophils and monocytes in the bone marrow. GATA-1, PU.1, and C/EBPs play critical roles in the transcriptional regulation of eosinophil lineage commitment and differentiation. Morphologic differentiation and maturation of the eosinophil parallel that of the neutrophil series, and its characteristic eosin-staining–specific granules are prominent by the myelocyte stage. IL-3, IL-5, and GM-CSF mediate eosinophil production in the marrow; IL-5, IL-13, chemokines (such as eotaxins [CCL11] regulated on activation, normal T cell expressed and secreted [RANTES; i.e., CCL5]), and leukotrienes [LTB4] play key roles in regulating eosinophil differentiation, chemotaxis, and functional activation.
After leaving the circulation, the majority of mature eosinophils reside in tissues, with a blood-to-tissue ratio estimated to be 1 : 300 to 1 : 500. The life span of tissue eosinophils is not known but may be several weeks, whereas the half-life in blood is around 24 hours. Eosinophils typically localize in areas exposed to the external environment, such as the tracheobronchial tree, gastrointestinal (GI) tract, mammary glands, and vagina and cervix. As discussed later, eosinophils have both immunoenhancing and immunosuppressive functions and play a role in helminthic infection, allergy, and the responses to certain tumors.
The mature eosinophil is slightly larger than the neutrophil, with a diameter of 12 to 17 µm. The nucleus is characteristically bilobed, although multiple lobes can be seen in patients with eosinophilia of diverse causes. The cytoplasm has prominent and morphologically distinctive granules that stain strongly with acid aniline dyes because of their high content of basic proteins.
Like the neutrophil, the mature eosinophil is endowed with the capacity for chemotaxis, phagocytosis, degranulation, and the synthesis of reactive oxidants and arachidonate metabolites. Eosinophils may also undergo a process similar to neutrophil netosis. Eosinophil cell surface membranes express a wide variety of molecules, including receptors for immunoglobulins and members of the immunoglobulin superfamily; cytokine receptors; adhesion molecules; chemokine, complement, and other chemotactic receptors; and major histocompatibility complex (MHC) class I and II and costimulatory molecules.
The distinctive mature eosinophil granules are membrane-bound organelles, 0.15 to 1.5 µm in length and 0.3 to 1 µm in width that contain a variety of enzymes and cytotoxic proteins. These eosin-staining specific granules are large ovoid bodies that contain an electron-dense crystalloid core surrounded by a less dense matrix. The eosinophil major basic protein (MPB) makes up about 50% of the dense crystalloid core of the eosinophilic specific granule. MBP from eosinophils induces histamine release from basophils and mast cells and also an autocrine degranulation of eosinophils. Release of proteins from these large granules is different from the degranulation process of neutrophils, where intact granules fuse with the surface membrane and empty their entire content and the membrane of granules is incorporated into the surface membrane. Instead, because the eosinophil is active in tissues, its granules swell and a tubulovesicular system extends from the granules to the surface permitting piece meal degranulation (PMD), or the graded release of granule content. Eosinophil peroxidase plays an important role in the antihelmintic function of eosinophils and utilizes bromate to generate hypobromous acid from hydrogen peroxide.
Much less common than eosinophil-specific granules are the primary granules characterized by their content of a lysophospholipase, the Charcot-Leyden crystal (CLC) protein that can polymerize to form the bipyramidal hexagons that are CLCs. CLCs are typically found in areas of eosinophil degeneration, such as sputum from asthmatic patients, nasal mucous of patients with allergies, stools of patients with parasitic infections, and the pleural fluid of patients with pulmonary eosinophilic infiltrates. The CLC lysophospholipase catalyzes the hydrolysis and inactivation of lysophospholipids generated by phospholipase A 2 , thus preventing the generation of proinflammatory arachidonic acid metabolites. The CLC protein composes about 5% of the total protein in eosinophils.
Slightly more numerous but distinctly smaller than primary granules are the small type granules whose content is not well characterized and secretory vesicles, which like secretory vesicles of neutrophils contain the NADPH oxidase flavocytochrome b558 , and albumin, indicating an origin as endocytic vesicles; however, in eosinophils these may also be part of the tubulovesicular system of PMD.
Eosinophil granule products, particularly MBP, eosinophil peroxidase, and eosinophil neurotoxin, are toxic to tissues, including the heart, lungs, and brain. These mediate many of the adverse clinical complications of eosinophilia and hypereosinophilic syndrome (HES), such as Löffler endocarditis and pneumonia.
Basophils and Mast Cells
Basophils and mast cells are believed to share a common progenitor cell, but mast cells leave the bone marrow to proliferate and mature in tissues, whereas full differentiation of basophils occurs in the bone marrow over 7 days before their release into the bloodstream; they are not normally found in the connective tissues. Basophils account for approximately 0.5% of the total circulating leukocytes and 0.3% of nucleated marrow cells. Mature basophils have a bilobed nucleus. Although basophils are distinctly smaller (5 to 8 µm) than mast cells (20 µm), they both contain large metachromatic granules that stain purple or bluish with Wright stain because of their high content of sulfated glycosaminoglycans. These granules are rich in heparin-type and chondroitin sulfate-type glucosaminoglycans linked to the serglycin protein backbone that is responsible for packing the cationic proteins, histamine, and kallikrein. Mast cells and basophils express high affinity receptor for the Fc portion of immunoglobulin E (IgE), which is an important trigger for release of granule contents and production of arachidonic acid metabolites in anaphylactic degranulation on their plasma membrane, and are key effector cells in certain hypersensitivity reactions. However, mast cells lack receptors for IL-2, IL-3, and CD11b/CD18 that are present on basophils. The heparin of basophils appears to have poor anticoagulant activity. Basophil granules also contain small amounts of MBP as well as serine proteases. Basophils synthesize and secrete IL-4 and IL-13 and may thus mediate a link between the innate and adaptive immune systems for generation of Th2 lymphocyte responses. Murine mast cells can secrete a wide variety of mitogenic or inflammatory cytokines (including IL-1, IL-3, IL-4, IL-5, and IL-6), chemokines, GM-CSF, and TNF-α, that are likely to play an important role in leukocyte recruitment and inflammation.
Mast cells are ordinarily distributed throughout normal connective tissue, where they are often situated adjacent to blood and lymphatic vessels, near or within nerve sheaths, and beneath epithelial surfaces that are exposed to environmental antigens such as the respiratory and GI tracts. The c-kit receptor for stem-cell factor is present on mast cells, but absent from the majority of basophils. The c-kit ligand, or stem-cell factor (SCF), is the main survival and developmental factor for mast cells, but other growth factors and cytokines such as IL-3, Il-4 and IL-9 are also supportive for mast cell development. Mature mast cells do not circulate in the blood, although circulating mast cell progenitors have been described and retain a limited proliferative capacity in the tissue compartment. In contrast to monocytes and macrophages, a transformation between the circulating and tissue forms of basophils and mast cells has not been observed.
Mast cells can be categorized into several types in mice and humans. In mice, the different types can be distinguished by their main tissue distribution and the major type of proteoglycan in their granules (heparin-containing mast cells are predominantly connective tissue and serosal mast cells, whereas chondroitin sulphate-containing mast cells are associated with mucosal surfaces). In humans two major subsets are identified based on the their content of chymase and tryptase. Both contain tryptase, but a subset that is particularly low in chymase largely secretes cytokines and chemokines and does not degranulate as opposed to the chymase rich type.
Mononuclear Phagocytes
The blood monocyte is derived from a bone marrow progenitor cell, the GMP, shared with the neutrophil, and undergoes differentiation through stages as monoblasts and promonocytes in the bone marrow. Monocytic differentiation of GMP is favored by a high level of the transcription factor PU.1 relative to C/EBPα, whereas the opposite drives granulocytic differention. The transit time for monocytes in the marrow compartment is briefer than for neutrophils, and the mature monocyte is released into the circulation only 24 hours after the last mitosis (see Table 22-1 ). Consequently, a relative monocytosis in peripheral blood commonly precedes the return of granulocytes during recovery from bone marrow aplasia or hypoplasia. Monocyte production and differentiation are regulated by IL-3, IL-6, GM-CSF, and the more lineage-specific cytokine M-CSF.
The monocyte may spend several days in the intravascular compartment in either circulating or marginated pools. Monocytes migrate into tissues and body cavities to participate in inflammatory processes as exudate macrophages and to replenish the resident tissue macrophage population, which has a relatively long life span. In patients receiving allogeneic bone marrow transplants, host tissue macrophages disappear gradually and are replaced by donor macrophages approximately 3 months after transplantation. Recent studies in mice suggest that at least some tissue macrophages are derived from stem cells that migrate to tissues during fetal development and maintain the macrophage pools in situ, provided these stem cells are not destroyed as occurs during allogeneic stem-cell transplantation as shown in mouse models.
The circulating monocyte in Wright-stained blood smears is 10 to 18 µm in diameter with a convoluted surface, a grey-blue cytoplasm, and an indented or kidney-shaped, foamy nucleus. However, some monocytes can be as small as 7 µm in diameter and difficult to distinguish morphologically from lymphocytes. Like neutrophils, monocytes contain secretory vesicles but only a single major class of granules with lysosomal characteristics, of which subsets have been described based on content of transforming growth factor α (TGF-α). Traditionally, two circulating populations of monocytes are identified by flow cytometry as either classical monocytes or the nonclassical monocytes. Classical monocytes have a high expression of CD14 (the LPS receptor) and a low expression of CD16 (the low-affinity immunoglobulin G [IgG] receptor), accounting for approximately 90% of circulating monocytes, whereas nonclassical monocytes have a high expression of CD16. An intermediate type, previously assigned to the nonclassical type, is now recognized as having high expression of CD14 and intermediate expression of CD16. These CD16 expressing types are in general seen as possible precursors of dendritic cells. The relative distribution among these subsets is not fixed and changes with exposure to growth factors and cytokines such as M-CSF and TNFα.
After leaving the circulation, monocytes become larger and take on the appearance of tissue macrophages characteristic of the organ in which they reside. The macrophage nucleus is typically oval with more prominent nucleoli, and the cytoplasm stains blue because of an increase in RNA content. Monocytes and macrophages are distinguished histochemically by the presence of a fluoride-inhibitable nonspecific esterase and can be identified immunohistologically by a variety of monoclonal antibodies such as F4/80 in the mouse and anti-CD68 in human tissue.
Monocytes and macrophages share many structural and functional features with neutrophils, and are capable of sensing chemotactic gradients, migrating to inflamed sites, ingesting microorganisms, and killing them using a variety of cytocidal products. However, compared to neutrophils, mononuclear phagocytes have a large and diverse developmental potential. In addition to their protective function as phagocytic cells in host defense, mononuclear phagocytes play a central role in the adaptive immune response by presenting antigens to lymphocytes, elaborate growth factors and cytokines important for lymphocyte function, wound repair, and hematopoiesis, and participate in a variety of scavenger and homeostatic pathways. Mononuclear phagocytes at inflammatory sites become “activated,” displaying morphologic alterations and a variety of enhanced functions. These include a more pronounced ruffling of the plasma membrane and pseudopod formation, an increased capacity for adherence and migration to chemotactic factors, increased microbicidal and tumoricidal activity, and enhanced ability to release cytokines.
Monocytes and tissue macrophages are considered to make up a “mononuclear-phagocyte system.” Resident tissue macrophages were formerly referred to as histiocytes, an imprecise and often loosely applied term. Tissue macrophages are widely distributed and perform specialized functions at portals of entry such as the pulmonary alveoli and in sterile sites such as the bone marrow.
Spleen
Macrophages are distributed in all parts of the spleen, including the germinal centers where they are associated with lymphocytes. Splenic macrophages located in the red pulp and sinuses serve a clearance function, where a sluggish blood circulation maximizes the interaction between blood elements and the macrophages lining the sinus walls.
Liver
The portal circulation percolates through a labyrinthine system, the spaces of Disse, before exiting via the hepatic venous system. This hepatic circulation, although less sluggish than that of the spleen, provides considerable contact between the blood and the resident liver macrophages, known as Kupffer cells, that reside within these vascular sinuses.
Lymph Nodes
As in the spleen, macrophages exist through all regions of peripheral lymph nodes. They are most abundant in the medullary zone close to efferent lymphatic and blood capillaries. This location is likely related to the important role macrophages play in the presentation of antigens to T lymphocytes.
Lungs
Pulmonary macrophages reside both in the interstitium of alveolar sacs and free within the air spaces, where they participate in the clearance of inhaled microorganisms and particulate matter. The number of lung macrophages increases in many chronic pulmonary inflammatory disorders. Pulmonary macrophages are easily seen in lungs of smokers, where black inclusions mark the macrophage vacuoles. Hemosiderin-laden alveolar macrophages can be indicative of recurrent pulmonary hemorrhage, such as in idiopathic hemosiderosis or Goodpasture syndrome. Gastric aspiration to detect ingested iron-laden macrophages is a useful test for these disorders.
Bone Marrow
Macrophages are found throughout the bone marrow cavity. They are particularly abundant within hematopoietic islands and on the walls of the marrow sinuses. Bone marrow macrophages may have a clearance function in normal or pathologic states of ineffective hematopoiesis. The clearance function of marrow macrophages is dramatically illustrated by the lysosomal storage diseases such as Gaucher disease. Large inclusions build up within marrow macrophages (as well as hepatic and splenic macrophages) because of the inability of these cells to break down lysosomal contents. Bone marrow macrophages also support hematopoiesis by modulating mesenchymal stem cells and osteoblasts to express retention signals, in particular CXCL12, for HSCs.
Other Sites
Mononuclear phagocytes associated with lymphoid cells reside throughout the alimentary tract, particularly in the submucosal tissues and small intestinal villi. They are present as microglial cells in the central nervous system (CNS), where their numbers increase after injury as monocytes emigrate across the blood-brain barrier, and they may contribute to the pathogenesis of the CNS manifestations of infection with the human immunodeficiency virus (HIV). Mammary gland macrophages released into milk during lactation have been implicated as a potential source of postnatal transmission of the HIV virus.
Dendritic Cells
Dendritic cells are specialized antigen-presenting cells with long cytoplasmic processes that are located in tissues throughout the body except for brain. They develop from a progenitor common with monocyte progenitors, supported by GM-CSF. M-CSF induces monocytic differentiation, and Flt3 ligand induces differentiation into a dendritic cell progenitor that may leave the bone marrow and home to lymphatic tissue and nonlymphoid tissue throughout life. In contrast, Langerhans cells of the epidermis, which are also antigen presenting cells, are self sustained and localize to skin during embryonic development. Some dendritic cells are also derived from the lymphoid lineage. Antigen presentation by dendritic cells, which have a high density of MHC class II molecules, is a particularly potent stimulus for T-cell mediation of the primary immune response. Plasmacytoid dendritic cells are a specialized population of dendritic cells, although derived from a shared precursor, that respond rapidly to viruses or nucleic-acid–containing complexes by secreting large amounts of type I interferon (IFN). NETs have been linked to activation of plasmacytoid dendritic cells and the pathogenesis of lupus.
Osteoclast
Osteoclasts are large, multinucleated mononuclear phagocytes that resorb mineralized cartilage and bone. Rodent transplantation studies have shown that osteoclasts can be derived from granulocyte-macrophage progenitor cells. Defects in osteoclast function result in osteopetrosis, a genetically heterogeneous group of disorders characterized by defective bone resorption. The Op/op osteopetrotic mouse mutant lacks M-CSF, which results in deficiencies of both osteoclasts and tissue macrophages. However, M-CSF levels and osteoclast numbers are normal in human infantile (“malignant”) osteopetrosis, an AR disorder with progressive obliteration of the marrow space. This severe form of osteopetrosis is caused by mutations in genes encoding a vacuolar proton pump or the chloride channel 7.
Function of Phagocytes
Phagocytic leukocytes play a central role in the acute phases of the inflammatory response, where they are rapidly mobilized into sites of tissue infection or injury and release an array of cytotoxic molecules to quickly eliminate the offending substance or microbe, as well as mediators that initiate an adaptive immune response. Phagocytes are also essential for normal repair of tissue injury, as evidenced by the impairment in wound healing in patients with deficits in leukocyte function or number.
The classic signs of the inflammatory response were described by the Roman writer Celsus as “rubor et tumor, cum calore et delore,” or, “redness and swelling with heat and pain.” However, it was not until the late nineteenth century that the cellular events associated with these signs were studied by Virchow and by Cohnheim. The beneficial role of phagocytes in the inflammatory process for host defense and wound healing was championed by Metchnikov. He received a Nobel prize for his work, much of which involved studies on the wandering ameboid mesenchymal cells of marine organisms like the larval starfish, for which he coined the term phagocyte, after the Greek word, phagein , “to eat.”
In this section, the principle functions of granulocytes and mononuclear phagocytes in the inflammatory process are reviewed. Although these functions will be discussed as individual components, it is important to recognize that many occur either simultaneously or in rapid succession. An overview of early events in the inflammatory process and cross talk between different leukocyte populations is shown in Figure 22-2 . Note that proinflammatory events that are critical for the response to tissue injury, and the effective elimination of microbial challenge also sets into motion the generation of counterregulatory signals leading to resolution of the inflammatory response.
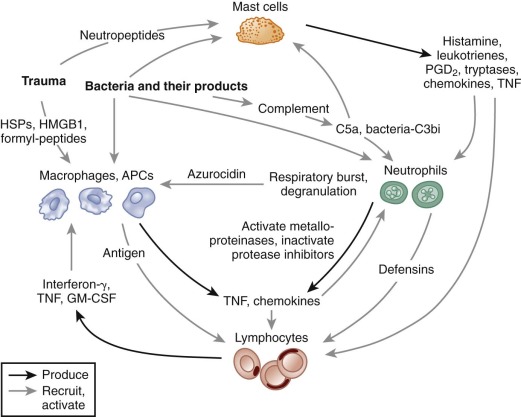
Humoral Mediators of the Inflammatory Response
The acute inflammatory response reflects an ongoing collaboration between tissue macrophages and mast cells, vascular endothelial cells, and circulating phagocytes. The release of soluble inflammatory mediators is crucial for activating and coordinating this process. These molecules can be generated from plasma proteins (e.g., the complement-derived protein fragment C5a), secreted by endothelial cells or inflammatory leukocytes (e.g., lipid metabolites, histamine, cytokines, S100 proteins), derived from invading microbes (e.g., endotoxin or formylated chemotactic peptides), or released from damaged cells (e.g., heat shock proteins [HSPs], the nuclear protein high molecular–group box 1 protein [HMGB1]).
The proinflammatory cytokines TNF-α and IL-1 have a broad range of activities in the acute inflammatory response. Both IL-1 and TNF can cause fever and muscle breakdown and are involved in the cachexia associated with chronic infection and malignancy. The synthesis of acute phase reactants by the liver is induced by IL-6, whose synthesis and secretion is stimulated by IL-1. Proinflammatory cytokines also induce a proadhesive state on the surface of endothelium and increase the production of the chemotactic cytokines (chemokines). IFN-γ is another important proinflammatory mediator that enhances the responsiveness of phagocytes to inflammatory stimuli. Counterbalancing the activities of these polypeptides are IL-4, IL-10, and TGF-β, which tend to down-regulate the acute inflammatory response.
Lipid mediators play both proinflammatory and antiinflammatory roles. As the inflammatory process progresses, a “class switch” is observed in neutrophils such that lipoxygenase activity induces production of antiinflammatory lipoxins instead of proinflammatory leukotrienes. In addition products of ω-3 unsaturated fatty acids, resolvins D1 and D2, are generated by neutrophils, and the related maresins are generated by efferocytosing macrophages. These products inhibit neutrophil transmigration by downregulating surface proteins involved in transepithelial migration, by inducing nitric oxide (NO) production in endothelial cells, and by enhancing the production of antiinflammatory cytokines and inhibiting production of proinflammatory cytokines of macrophages and making them more apt for uptake of apoptotic neutrophils.
Vasodilation and increased vascular permeability are two early responses to an inflammatory insult that are elicited in large part by products secreted by granulocytes and mononuclear phagocytes. Activated basophils and tissue mast cells release histamine, which leads to vasodilation of tissue arterioles and microvascular beds through H 1 -type receptors. The lipid metabolite platelet activating factor (PAF), which is secreted by activated macrophages, mast cells, and endothelial cells, induces platelet degranulation and the release of additional histamine and also serotonin, another vasoactive amine. Prostaglandin E and other arachidonic acid metabolites secreted by activated neutrophils and macrophages are another group of potent vasodilators. Finally, vasodilation can be triggered by the release of NO from endothelial and smooth muscle cells as well as perhaps from activated macrophages, which may be particularly important in the hypotension seen with gram-negative septicemia. The increased vascular permeability that produces the edema of acute inflammation allows plasma proteins such as immunoglobulins and complement to enter tissues to promote phagocyte activation and opsonize microbes. Agents that increase vascular permeability include histamine, serotonin, PAF, and leukotrienes (LTs) C 4 , D 4 , and E 4 . Bradykinin, which is generated as the result of Hageman factor (factor XII) cleavage, also induces enhanced vascular permeability.
Chemokines, Small Lipids and Other Chemoattractants
A wide variety of chemoattractants for neutrophils and other circulating leukocytes are generated at sites of inflammation ( Table 22-4 ). These molecules are chemically diverse and are derived from many different sources in response to bacterial products and inflammatory mediators released as a result of tissue necrosis. This diversity provides a functional redundancy and ensures that leukocytes will be attracted to sites of injury or infection. In addition to their role as chemoattractants, the molecules listed in Table 22-4 induce the activation of many other phagocyte functions upon binding to their cognate cell-surface receptors. These include the upregulation and increased affinity of leukocyte integrin adhesion receptors to promote firm attachment to the endothelium, degranulation, and activation of the phagocyte respiratory burst. Many chemoattractants are secreted by activated phagocytes, which act as a positive feedback loop for additional recruitment and activation of inflammatory cells.
| Chemoattractant | Receptor(s) | Source | Upregulators | Target Cells |
|---|---|---|---|---|
| Lipids | ||||
| PAF | PAFR | N, E, B, P, M, endothelium (phosphatidylcholine metabolism) | Calcium ionophores | N, E |
| LTB 4 | B-LTR | N, M (arachidonate metabolism) | Microbial pathogens, N -formyl peptides | N, M, E |
| 12-HETE | P (arachidonate metabolism) | Platelet activation | E | |
| CXC Chemokines | ||||
| IL-8 (CXCL8) | CXCR1, 2 | M, N, endothelium, many other cells | LPS, IL-1, TNF, IL-3 | N, B |
| GRO α, β, γ (CXCL1, 2, 3) | CXCR2, 1 | M, endothelium, many other cells | IL-1, TNF | N, B |
| NAP-2 (CXCL7) | CXCR2 | P * | Platelet activators | N |
| PF4 (CXCL4) | CXCR3B | P | Platelet activators | N, M, E |
| SDF-1 (CXCL12) | CXCR4 | Marrow stroma, other | N, M, B, T | |
| Fractalkine (CX 3 CL1) | CX 3 CRI | M, endothelium, other | IL-1, TNF, LPS, IFN-γ | M, T, NK |
| CC Chemokines | ||||
| MCP-1, 2, 3, 4 | CCR2, 3 | |||
| (CCL2, 8, 7, 13) | M, endothelium, many other cells | IL-1, TNF, LPS, PDGF | M, B, E, T | |
| RANTES (CCL5) | CCR1, 3, 5 | M, E | IL-1, TNF, anti-CD3 | M, B, E, T |
| Eotaxin (CCL11) | CCR3 | M, endothelium, other | Allergens | E, B, TH2 |
| Other | ||||
| N -formyl peptides | fMLPR | Bacteria, mitochondria | — | N, M, E, B |
| C5a | C5aR | Plasma complement | Complement activation | N, M, E, B |
| PDGF | PDGFR | P | Platelet activation | M |
| TGF-β | TGFR | P, other | Platelet activation | N, M |
* Platelets, when activated, secrete platelet basic protein (PBP) and connective tissue–activating peptide III (CTAP-III), which are cleaved to NAP-2 by cathepsin.
The phospholipid PAF, released by both activated phagocytes and endothelial cells, triggers platelet activation and granule release in addition to being a potent chemoattractant for neutrophils and eosinophils. Activation of phagocytes also stimulates the phospholipase A 2 -mediated cleavage of membrane phospholipids to generate arachidonic acid, which is then converted into a variety of eicosanoid metabolites, including the chemoattractant leukotriene B 4 (LTB 4 ).
Chemokines (named for their combined chemo tactic and cyto kine properties) are a family of small (8 to 10 kDa) basic heparin-binding proteins that comprise an important group of phagocyte chemoattractants. Chemokines were first discovered in the late 1980s as molecules that interact relatively specifically with subsets of inflammatory leukocytes and therefore help orchestrate the sequential influx of neutrophils, monocytes, and finally lymphocytes into an inflamed tissue site. Proteoglycans on endothelial cells or in the subendothelial matrix bind chemokines to produce locally high chemokine concentrations at an inflamed site. As additional chemokines and their receptors have been identified, many other functions have emerged, including regulation of lymphoid homeostasis, hematopoiesis, and angiogenesis. Of note, SDF-1 (CXCl2) , provides a key retention signal for neutrophils in the marrow through its interaction with the CXCR4 receptor, and mutations in the CXCR4 receptor account for the WHIM syndrome, an inherited neutropenia.
Members of the chemokine family, which have a conserved structure containing two cysteine pairs, have been divided into two groups based on the disulfide sequence pattern. The CXC family, in which the first cysteine pair is separated by an intervening amino acid, include IL-8 (CXCL8), the growth-regulated oncogene (GRO) peptides ( CXCL1, 2, and 3 ), and neutrophil-activating protein 2 (NAP-2; CXCL7 ), which are all potent neutrophil activators and chemoattractants. The IL-8 and GRO chemokines are secreted by phagocytes and mesenchymal cells (including endothelial cells) in response to inflammatory mediators such as IL-1 and TNF. Fractalkine (CX 3 CL1) is unique in having three intervening amino acids between the first two cysteine residues. In addition, rather than being soluble, fractalkine is expressed on the cell surface, because it is linked via a mucinlike stalk to a transmembrane domain. The other major family of chemokines is called the CC family, because the first two cysteines are adjacent to each other. CC chemokines include two important inducers of mononuclear phagocyte migration, monocyte chemotactic protein 1 (MCP-1; CCL2 ) and RANTES (CCL5). MCP-1 is produced by a wide variety of cells, whereas RANTES is secreted by macrophages and eosinophils. RANTES is chemotactic for eosinophils, basophils, and memory T cells as well as monocytes, and both MCP-1 and RANTES induce histamine release from basophils.
Despite the diverse chemical structures of phagocyte chemoattractants listed in Table 22-4 , the corresponding receptors all belong to the seven-transmembrane–spanning receptor (7-TMR) family, also known as heptahelical or serpentine receptors, that are coupled to heterotrimeric G proteins. For chemokines, more than six receptors for CXC chemokines and ten receptors for CC chemokines have been identified. Most chemokines bind to more than one receptor, and most chemokine receptors, particularly those for CC chemokines, recognize more than one chemokine. Neutrophils, monocyte/macrophages, eosinophils, basophils, dendritic cells, lymphocytes, and T cells each express a distinctive subset of chemokine receptors. According to one model, specific receptors are used sequentially in successive gradients of chemoattractants. Some transmit desensitizing, rather than activating, signals or even fail to signal and act instead as “decoy” receptors to downregulate inflammatory reactions. Of note, a number of chemokine receptors are coreceptors for HIV-1, including CCR5 and CCR3, whose ligands include RANTES, and CXCR4, the major receptor for stromal cell–derived factor 1 (SDF-1), which is a chemoattractant for T lymphocytes, CD34+ hematopoietic progenitor cells, and neutrophils.
The signaling through 7-TMR has proven much more complicated (and powerful) than the original signaling mediated via associated heterotrimeric G proteins, which bind to specific intracellular domains of the receptor. Ligand binding to the receptor promotes the exchange of GTP for guanosine diphosphate (GDP) bound to the G protein α subunit, which in turns leads to the dissociation of the β-γ subunits and their interaction with downstream signaling effectors. In addition, 7-TMR associates with β arrestins and signals through these in a G-protein independent way. Finally, 7-TMR may transactivate tyrosine kinase growth factor receptors. Signals generated from 7-TMR activate enzymes that catalyze the production of important phospholipid second messengers at the cell membrane.
Adhesion and Migration into Tissues
The discovery that leukocytes migrate from the bloodstream into extravascular sites of inflammation, described by Cohnheim in 1867, was a major milestone in the conceptualization of the inflammatory process. Cohnheim, who used intravital microscopy to study the microvasculature in the frog tongue and mesentery after tissue injury, also first proposed that inflammatory stimuli induce a molecular change in the blood vessel wall that promoted the increased adherence of leukocytes, a concept that was finally proven a century later.
To move from the bloodstream into inflamed sites, leukocytes must attach to the vascular endothelium, migrate between adjacent endothelial cells in a process referred to as diapedesis, and penetrate the basement membrane. The molecular mechanisms underlying these events involve a series of sequential adhesive interactions between chemoattractant-activated leukocytes and endothelial cells that are activated by inflammatory mediators ( Fig. 22-3 ).
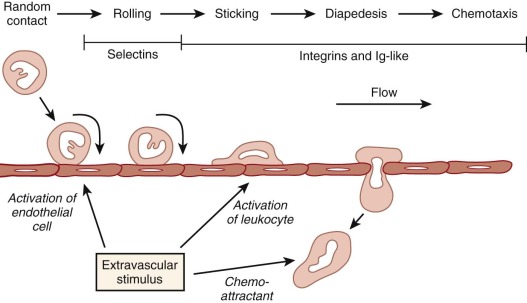
Leukocyte Adhesion and Migration into Tissues
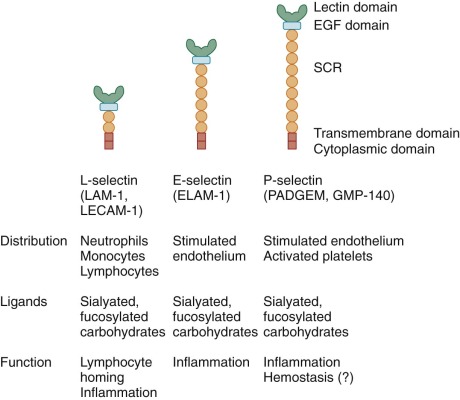
The initial step in emigration from postcapillary venules is a low-affinity interaction between the neutrophil and the endothelium that is often referred to as rolling because of its appearance in intravital microscopy. This transient adherence, also called tethering, is mediated by the upregulation of selectin expression on endothelial cells. The selectin family of adhesion molecules are membrane-spanning glycoproteins ( Fig. 22-4 ) that bind to fucosylated structures such as Lewis X (Galβ1→4 [Fucα1→3] GlcNac→R), Sialyl-Lewis X, and other specific carbohydrates. P-selectin is important for the initial steps of neutrophil adhesion to the endothelium and is stored in the Weibel-Palade bodies and α granules of endothelial cells and platelets, respectively. Upon endothelial cell activation by histamine, thrombin, and other inflammatory molecules, these cytoplasmic storage granules fuse with the cell membrane to rapidly increase the surface expression of P-selectin. E-selectin is expressed on endothelial cells at low levels, but it is upregulated by transcriptional activation and de novo protein synthesis in response to inflammatory cytokines. E-selectin binds to three different ligands on neutrophils: P-selectin glycoprotein ligand (PSGL) 1, E-selectin ligand (ESL) 1, and CD44. These ligands allow endothelial cells to capture neutrophils by mediating tethering, rolling, and slowing of neutrophil velocity, respectively. L-selectin is expressed constitutively on the surface of neutrophils, mononuclear phagocytes, and lymphocytes and is shed within minutes of leukocyte activation by a proteolytic cleavage event near the external membrane surface insertion site. Circulating L-selectin may modulate leukocyte adhesion during inflammation. PSGL-1 is constitutively expressed on the tip of microvilli on the neutrophil surface and remains associated during activation but moves to the uropod when the neutrophil polarizes.
Rolling neutrophils can detach and return to the circulation. Others will come to a halt and within seconds adopt a flattened, adherent morphology and attach firmly to the vessel wall. This firm attachment appears in large part to be mediated by leukocyte integrin adhesion receptors binding to intracellular adhesion molecules (ICAMs) on the endothelium. In addition, complement fragments are found on the endothelial surface at inflamed sites and may also function as integrin binding sites. Leukocyte activation by chemoattractants and other inflammatory mediators is critical to the development of these strong adhesive interactions, because it leads to the upregulation of the number and avidity of cell-surface integrins (“inside-out signaling”). Exposure to locally high concentrations of chemoattractants may be enhanced by selectin-mediated tethering and by the retention of chemokines on extracellular matrix.
The integrins are a large family of adhesion proteins that are glycosylated heterodimers of a noncovalently linked α chain and β chain and are classified into subfamilies according to the type of β subunit. Many integrins mediate attachment to extracellular matrices by serving as receptors for matrix proteins. Others are involved in hemostasis, such as glycoprotein IIb/IIIa on platelets. Neutrophil β 2 and β 1 integrins appear to be involved in regulating neutrophil retention and release, respectively, from the bone marrow storage pool into the circulation.
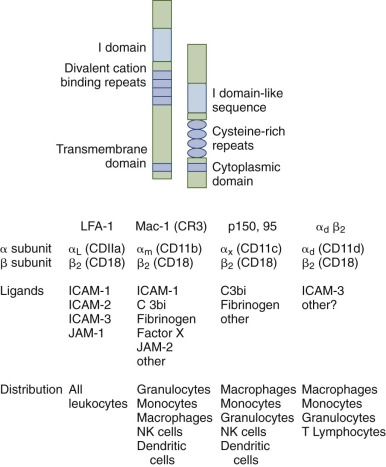
The leukocyte β 2 integrins ( Fig. 22-5 ) play a critical role in mediating adhesive interactions in inflammation, including the attachment of leukocytes to endothelial cells and are also opsonic receptors for complement fragment C3bi-coated particles. There are four different leukocyte β 2 integrins, each having a common 95 kDa β subunit (CD18) but different α subunits, CD11a (177 kDa), CD11b (165 kDa), CD11c (150 kDa), and CD11d (160kDa) (see Fig. 22-5 ). Lymphocyte function antigen 1 (LFA-1) is expressed on the surface of all leukocytes, including lymphocytes. Mac-1 and p150,95 are expressed by granulocytes, mononuclear phagocytes, some activated T lymphocytes, and large granular lymphocytes. Mac-1 is the most prominent β 2 integrin on neutrophils, whereas α d β 2 is expressed particularly in tissue macrophages. Mutations in the common β 2 subunit result in an inherited defect in phagocyte function, leukocyte adhesion deficiency type I (LAD I) as discussed in a later section. All β 2 integrins are absent in LAD I, indicating that the stability of each α subunit requires association with the β 2 chain. The β 2 subunit has a large, glycosylated extracellular domain, a single transmembrane-spanning domain, and a short cytoplasmic tail. The extracellular domain has two regions that are conserved among other β subunits. There are four cysteine-rich tandem repeats that appear to be important for the tertiary structure of the β subunit. Another conserved region, located near the N-terminus, is critical for maintenance of the α/β heterodimer and may also bind divalent cations. The α subunit is also a glycosylated integral membrane protein with a single membrane-spanning segment and a short cytoplasmic tail. The external domain contains three divalent cation-binding motifs that must be occupied for ligand binding to occur. A second important extracellular domain, the I domain (for inserted or interactive domain), can coordinate divalent cations and is also thought to be involved in ligand binding. The intracellular domain of the α subunit includes a conserved sequence that is critical for the modulation of integrin avidity (see “Leukocyte Adhesion Deficiency”).
Although β 2 integrins are constitutively expressed on the neutrophil cell surface, a large pool of Mac-1 is stored in intracellular secretory vesicles (see Table 22-2 ). These vesicles are rapidly mobilized upon neutrophil activation by chemoattractants and fuse with the membrane to increase the cell surface expression of β 2 integrins by about tenfold. Signaling through chemoattractant receptors also markedly increases the avidity of β 2 integrins for their ligands, which plays an even more important role in rapidly upregulating integrin activity and promoting firm attachment to the blood vessel wall. Integrins on inactivated cells have a bent position that does not allow ligand binding. Signals generated from 7-TMRs recruit talins to the cytoplasmic part of the integrin β chains, twisting the α and β chains apart and tethering the integrins to the actin cytoskeleton. This is assisted by actin-binding protein (ABP) 1 and by the protein Kindlin 3, which is only expressed in cells of hematopoietic origin. This changes the conformation of the extracellular parts of the αβ heterodimer to an extended form, now capable of ligand binding and hence also of transducing signals from outside in. Defects in Kindlin 3 results in LAD III (see “Leukocyte Adhesion Deficiency Type III”). Ligand binding to integrins results in clustering of the integrins and the transmembrane calcium channel Orai1 that mediates store operated calcium entry and activation of tyrosine kinase and other signaling cascades, which in addition to mediating adhesion, provide important costimulatory signals to enhance migration, respiratory burst, Fcγ-mediated phagocytosis, and degranulation.
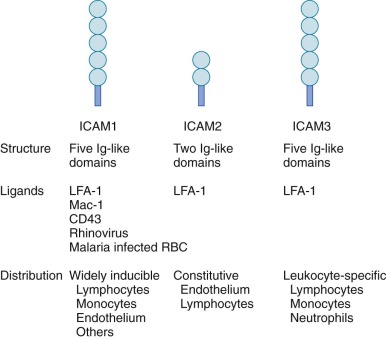
The major counterreceptors for the β 2 integrins are the ICAMs ( Fig. 22-6 ), which are members of the immunoglobulin superfamily. These transmembrane proteins contain anywhere from two to six immunoglobulin domains and are present on endothelial cells, T cells, and a variety of other cell types. ICAM-1 and ICAM-2 are of particular importance in mediating binding of neutrophils and other leukocytes to the endothelium. Endothelial cell expression of ICAM-1 increases in response to inflammatory cytokines, which promotes increased cell–cell interactions with leukocytes at inflamed sites. VCAM-1 is another immunoglobulin superfamily member expressed on endothelial cells that is inducible by cytokines. VCAM-1 is the counter-receptor for the β 1 integrin, VLA-4, and appears to be important in promoting the adherence of monocytes and eosinophils during inflammation. The β 2 integrin Mac-1 also has an important role as an opsonic receptor for the complement fragment, C3bi (see “Recognition, Opsonization, and Phagocytosis”).
Although leukocyte β 2 integrin-mediated adhesion is clearly important for neutrophil recruitment from the systemic microvasculature into inflammatory sites, neutrophil emigration out of the pulmonary circulation can also be mediated by alternative pathways, depending on the inflammatory stimulus. Whether the alternative pathway in pulmonary capillaries, which are in close proximity to the pulmonary epithelial cells, involves selectins or other adhesion molecules remains to be defined. TREM-1 is essential for pulmonary transepithelial migration of neutrophils.
The final steps in emigration of neutrophils from the blood vessel lumen into inflamed tissue involves squeezing between adjacent endothelial cells (diapedesis) and penetrating the basement membrane (see Fig. 22-3 ). The presence of a chemotactic gradient is required to induce the directional migration of neutrophils. Adhesive interactions between the β 2 integrins and endothelial cell ICAM-1 are essential for neutrophil diapedesis, whereas VCAM-1 and E-selectin can mediate the transmigration of monocytes and eosinophils. Junctional adhesion molecule (JAM) 1, an immunoglobulin superfamily protein that is expressed at tight junctions of resting endothelial cells and epithelial cells, facilitates leukocyte transmigration via binding to LFA-1 (CD11a/CD18). The tight junction between endothelial cells is weakened by loss of paxillin and focal adhesion kinase in proximity to migrating neutrophils. Transendothelial migration of neutrophils is also dependent on homologous binding between neutrophil and endothelial cell platelet–endothelial cell adhesion molecule (PECAM) 1 (CD31), another immunoglobulin superfamily member expressed on the surface of leukocytes, platelets, and endothelial cells, where it is localized at the junctions between cells. Signals from ICAM-1 induce vascular permeability, activate PECAM-1 for enhanced adhesivity, and support of neutrophil transendothelial migration. The JAM-C expressed by endothelial cells seems important for directing neutrophil migration into tissues and inhibiting migration in the reverse direction. Migrating neutrophils induce increases in endothelial intracellular calcium levels and changes in actin cytoskeleton that facilitates transmigration. To find gaps between pericytes sheathing endothelial cells, neutrophils use ICAM-1 to crawl along pericyte extensions, which further guide their migration into tissues. Finally, chemoattractant-induced neutrophil degranulation results in the release of digestive enzymes, including collagenase, elastase, and gelatinase, which may facilitate basement membrane penetration.
Chemotaxis
Chemotaxis is the directional movement of a cell along a concentration gradient. Defects in neutrophil cellular motility or other steps in chemotaxis can result in decreased resistance to bacterial and fungal infections, as discussed later in this chapter. Cells respond to a chemotactic gradient by sensing constantly across their surface, and bound chemotactic receptors are continuously internalized. A migrating neutrophil has a polarized appearance, extending pseudopodia or lamellipodia, thin structures rich in actin filaments and lacking intracellular organelles, at the leading edge. The pseudopods appear to glide forward, pulling the cell body behind them. The nucleus tends to remain at the posterior half of the moving leukocyte. Migration also requires formation of a uropod at the “tail” of the leukocyte, which detaches from the underlying matrix and retracts the rear of the cell as it moves forward. Rho GTPases play an important role in establishing chemoattractant-induced polarization and migration. Ly49Q, a surface-expressed MHC I–associated receptor is associated with the SH2 domain–containing protein tyrosine phosphatase (SHP) 1 phosphatase in nonpolarized neutrophils but exchanges SHP-1 with SHP-2 in activated cells, which results in reorganization of lipid rafts and polarization.
Neutrophil movement is dependent on the dynamic assembly and disassembly of filamentous actin, which is coordinated by various ABPs whose activity is regulated by intracellular signaling molecules. Actin can exist as either a soluble monomer (globular actin) or in needlelike helical filaments (filamentous actin). Actin filaments align spontaneously in parallel bundles, but in the cell are organized into a branching network because of the presence of actin filament cross-linkers such as ABP. Agonists acting via receptors on the cell membrane trigger the generation of second messengers, which interact with ABPs to control dynamic local cycles of filamentous actin assembly. The Rho GTPases (Cdc42 and Rac) are important regulators of actin remodeling and are activated by ligand binding to chemoattractant receptors. Cdc42 activates proteins of the Wiskott-Aldrich syndrome protein (WASP) family, which then bind to a complex of seven proteins known as the Arp2/3 complex to nucleate assembly of new actin filaments at the leading edge of migrating cells.
During pseudopod extension in chemoattractant-activated neutrophils, new actin polymerization occurs at the site of membrane protrusion while the filamentous actin in the rear of the cell disassembles. How actin assembly–disassembly results in membrane extension and formation of pseudopodia is not fully understood, but it may involve localized changes in osmotic pressure caused by changes in actin polymerization. Membrane movement and retraction at the rear of the cell is mediated in part by Rho GTPase-regulated contractile proteins such as myosin 1.
Recognition, Opsonization, and Phagocytosis
The recognition, ingestion, and disposal of microbes, foreign particulate matter, and damaged cells constitutes a major aspect of phagocyte function. To facilitate their recognition by phagocytes, these targets are coated with serum opsonins (the term opsonin is from Greek, to prepare for dining ) that include proteolytic fragments derived from the complement cascade as well as specific immunoglobulins. The key humoral opsonins are the proteolytic cleavage products of C3 (C3b and C3bi), which can be generated in the absence of specific immunity by the alternative and mannose-binding lectin pathways (see later), and the opsonic antibodies, IgM, IgG 1 , and IgG 3 . Targets are opsonized by the deposition of C3b and C3bi or of IgG onto their surfaces via the specific (Fab) portion of the antibody. Antibacterial IgM antibodies, although not opsonic by themselves, play an important role in phagocytosis by activating complement. Opsonins are recognized by phagocyte cell-surface glycoprotein receptors for immunoglobulin and C3 cleavage products, as described later. Inherited deficiencies of opsonization can result in increased susceptibility to bacterial infections, as discussed in “Disorders of Chemotaxis.” In contrast, primary defects in phagocyte receptors for these opsonins appear to be an uncommon cause of recurrent infections.
Phagocytes also have cell surface receptors capable of recognizing targets even in the absence of opsonins. These members of the pattern recognition receptor (PRR) family recognize broad classes of macromolecules. PRRs recognize molecular signatures unique to microbes, known as PAMPs (such as LPS or molecules released from damaged or stressed tissue known as DAMPs), such as adenosine triphosphate, as discussed previously in this section. PRRs include scavenger receptors, which have broad binding specificity for polyanionic ligands and participate in the clearance of diverse materials, including modified low-density lipoprotein (LDL) and apoptotic cells. The mannose receptor recognizes carbohydrate structures present in a range of bacteria, fungi, virus-infected cells, and parasites whereas the β-glucan receptor (dectin-1) binds to β-glucan structures in zymosan and other yeast-derived particles. Mammalian TLRs are an important group in the PRR family. At least 12 different TLRs have been described, which recognize conserved peptide, lipid, carbohydrate, and nucleic-acid structures expressed by different groups of microbes. For example, peptidoglycan and LPS are PAMPs associated with gram-positive and gram-negative bacteria and are recognized by TLR-2 and TLR-4, respectively. Binding to PRRs, which often occurs in a combinatorial fashion, activates an array of proinflammatory responses including expression of proinflammatory cytokines and release of oxidants and reactive nitrogen intermediates. TLR signaling activates the nuclear factor κB (NF-κΒ)– and IFN-regulatory factor (IRF)–dependent pathways, and genetic defects in the protein that couples TLRs to NF-κΒ lead to recurrent bacterial infections. Some PRRs also trigger phagocytosis, including the mannose receptor, scavenger receptors, and β-glucan receptor.
In addition to cell surface PRRs, soluble PRRs present in serum or tissues serve important roles in alerting phagocytes to the presence of microbes. These include the collectins (calcium-dependent lectins) that bind to oligosaccharide or lipid moieties of microorganisms to enhance their opsonization and efficiently activate phagocytes. Members of the collectin family include the lung alveolar surfactant proteins A and D and the mannose-binding lectin (MBL), also known as mannose binding protein. The binding of MBL to mannose residues initiates a third pathway of complement activation (in addition to the classical and alternative pathways) by interacting with the MBL-associated serine proteases (MASPs) 1, 2, and 3, which generates opsonic C3 fragments. MBL can also function itself as an opsonin for promoting uptake by the CR1 complement receptor. Ficolins 1, 2, and 3 are other soluble PRRs that are capable of recruiting MASPs. Pentraxin 3 is a soluble pattern recognition molecule stored in neutrophil-specific granules and important for humoral innate immunity.
Inflammasomes
Signaling through the membrane-bound PRRs in the previous paragraph generally leads to activation of the NF-κB pathway or IRF and production of proinflammatory cytokines, in particular IL-1β and IL-18 and type-1 IFNs, respectively. IL-1β and IL-18 are both without signal peptides and stored in the cytosol as proforms, but when processed by proteases, which in turn are activated by inflammasomes, these cytosolic cytokines are secreted from the cells by unconventional secretion mechanisms. IL-1β and IL-18 play a major role in activation of several steps of the inflammatory process, including endothelial cell and B-cell activation. Inflammasomes are structures that are assembled from preformed subunits in the cytosol in response to a variety of stimuli, such as signaling through cell surface (as discussed) or intracellular PRR such as the nucleotide-binding oligomerization domain–like receptors (NLRs) and retinoic acid–inducible gene 1–like receptor. Depending on the activating stimulus, different subunits become engaged in formation of the macromolecular complex known as an inflammasome and activate procaspase 1 to process the cytosolic proIL-1β and proIL-18 to the mature forms for secretion. A major cytosolic protein of neutrophils, pyrin, forms part of some inflammasomes, and mutations in the pyrin gene MEVF cause the inflammatory condition known as familial Mediterranean fever.
Complement Receptors
The activation of complement C3 to generate the cleavage fragments C3b and C3bi is the dominant source of opsonins in the absence of antibodies. Four C3 fragment receptors have been described on phagocytic leukocytes ( Table 22-5 ). The human phagocyte C3b receptor (CR1; CD35) is a high–molecular-weight, single-subunit glycoprotein that shows a substantial heterogeneity in size because of the presence of four distinct alleles in the human population that encode proteins ranging in size from 160 to 250 kDa. CR1 is responsible for the binding of C3b-opsonized particles and for initiating their ingestion. CR1 also recognizes microbes opsonized with MBL. The other major opsonic receptor, CR3, recognizes particles opsonized with C3bi. This receptor is the same as the β 2 integrin Mac-1 (CD11b/CD18) discussed in detail in an earlier section (see Fig. 22-5 ). Binding of C3bi-opsonized particles to Mac-1 triggers both phagocytosis and, in neutrophils, the respiratory burst. The CR4 receptor is the same as the β 2 integrin CD11c/CD18 and, on macrophages, can also initiate phagocytosis of C3bi-coated targets. Finally, a newly discovered receptor that binds the complement fragments C3b and C3bi, termed complement receptor of the immunoglobulin superfamily (CRIg) is expressed primarily in resident macrophages of the liver (Kupffer cells). CRIg appears to play a critical role in mediating phagocytosis and clearance of C3 fragment-opsonized bacteria from the circulation, at least in mice.
| CR1 (CD35) | CR3 (CD11b/CD18; Mac-1) | CR4 (CD11d/CD18) | CRIg | |
|---|---|---|---|---|
| Cell distribution | Neutrophils Monocytes B lymphocytes | Neutrophils Monocytes Macrophages NK cells | Macrophages Dendritic cells | Kupffer cells Certain other resident tissue macrophages |
| Structure | Transmembrane protein | Transmembrane, two subunits | Transmembrane, two subunits | Transmembrane protein |
| Ligands | C3b C4b C3bi C1q MBL (mannose-binding protein) | C3bi ICAM-1, 2 Fibrinogen Factor X | C3bi Fibrinogen | C3b C3bi |
| Function | Phagocytosis | Phagocytosis Respiratory burst (neutrophils) Adhesion Activation | Phagocytosis Adhesion | Phagocytosis |
Immunoglobulin Receptors
Immunoglobulins are recognized by Fc receptors (FcRs), which are members of the immunoglobulin gene superfamily. FcRs bind to the “constant domain” of the antibody molecule, these being specific to each class of immunoglobulins (IgA, IgE, IgG, and IgM). The most important from the standpoint of microbial opsonization are those FcRs that recognize IgG (FCγRs) ( Table 22-6 ). The FcγRs include three distinct classes, FcγRI, FcγRII, and FcγRIII, which are encoded by at least eight genes that have evolved through gene duplication and alternative splicing, although not all give rise to detectable mRNA and/or protein. The low-affinity FcγR genes (FcγRII and III families) and two of the genes for the high-affinity IgE receptor are clustered on chromosome 1q22. The high-affinity FcγRs map to other sites on chromosome 1.
| FcγRIa (CD64) | FcγRIIa (CD32) | FcγRIIIa, b (CD16) | |
|---|---|---|---|
| Polymorphisms | 131R/H | IIIa: 48L/R/H, 158F/V IIIb: NA-1, NA-2 | |
| Cell distribution | Monocytes Macrophages IFN-γ– or G-CSF–treated neutrophils IFN-γ–treated eosinophils | Monocytes Neutrophils Macrophages Eosinophils Basophils Platelets | IIIa: macrophages, monocytes (some), T cells, NK cells IIIb: neutrophils, IFN-γ–treated eosinophils |
| Protein size, type | 72 kD, transmembrane | 40 kD, transmembrane | 50-90 kD IIIa: transmembrane IIIb: GPI anchored |
| Associated proteins | FcRγ homodimer | — | IIIa: FcRγ homodimer (macrophage, monocyte) IIIb: colligation with FcγRIIa or CR3 |
| Ligands | Monomeric IgG (G1 = G3 > G4 > G2) | Complexed IgG (G1 = G3 > G2, G4) (FcγRIIa 131H-G2) | Complexed IgG (G1 = G3 > G2, G4) |
| Affinity | High | Low | Low |
| Function | Phagocytosis Respiratory burst Endocytosis of IC ADCC Antigen presentation | Phagocytosis Respiratory burst Exocytosis Endocytosis of IC ADCC Antigen presentation | Phagocytosis Exocytosis Endocytosis of IC ADCC (IIIa) Antigen presentation (IIIa) |
Except for FcγRIIIb, which is anchored in the membrane by a GPI moiety, the FcγR proteins have a single transmembrane domain. FcγRI and FcγRIIIa exist as oligomeric complexes with γ (in phagocytes) or ζ (in lymphocytes) chains that are important both for their stable expression and signaling functions. These accessory chains contain YXXL ITAM motifs that become tyrosine phosphorylated upon receptor cross-linking to initiate downstream signaling cascades. The cytoplasmic tail of the FcγRIIa receptor contains variant ITAM motifs, and hence this receptor does not require accessory chains for its function. There is no murine equivalent to the FcγRIIa receptor. The GPI-linked FcγRIIIb receptor must be co-ligated to either FcγRIIa or the β 2 integrin CR3 (Mac-1) to initiate signaling functions upon ligation. FcγRIIb receptors, expressed on monocyte/macrophages, mast cells, and lymphocytes, contain ITIM sequences and instead inhibit cellular activation upon immunoglobulin cross-linking.
Each FcγR is expressed at different levels, depending on the type of phagocytic cell (see Table 22-6 ), and some FcγRII and FcγRIII family members are also expressed on lymphocytes, platelets, thymocytes, natural killer (NK) cells, and mast cells. The FcγR1 class includes the products of three highly homologous genes, denoted A, B, and C, although the protein products of the latter two have not been detected in vivo. The FcγR1a receptors are expressed by monocytes and neutrophils or eosinophils stimulated by IFN-γ or G-CSF and have a high affinity for monomeric IgG. Members of the FcγRII and FcγRIII families have a low affinity for monomeric IgG but a high affinity for clusters of IgG (e.g., several antibodies bound to the same particle) and immune complexes. Only neutrophils and IFN-γ–stimulated eosinophils appear to express FcγRIIIb, and polymorphisms in this receptor correspond to the serologically defined NA1/NA2 antigen system, which is a common antibody target in alloimmune and autoimmune neutropenia (AIN). Polymorphisms in FcγRIIa, FcγRIIIa, and FcγRIIIb are associated with an increased incidence or severity of various autoimmune or infectious diseases. For example, the 131H allele of FcγRIIa, which is the only FcγR that mediates efficient phagocytosis of IgG 2 -opsonized bacteria, has been linked to a lower incidence of infections with encapsulated bacteria.
Cross-linking of FcγRs can trigger a wide range of functional responses, including phagocytosis of IgG-coated microbes, blood cells, or tumor cells; ingestion of immune complexes; release of inflammatory mediators; activation of the respiratory burst; and antibody-dependent cellular cytotoxicity (ADCC). The relative roles of different phagocyte FcγR classes have not been clearly delineated, although all can function as receptors for opsonized particles or immune complexes. Binding of IgG to FcγRI and FcγRIIa activates the respiratory burst, whereas secretion of granular contents is a prominent response upon binding to FcγRII and FcγRIIIb. Although the ligand-binding domains of different FcγRs all bind IgG, the specificity of the cellular response may be governed by the unique transmembrane and cytoplasmic domains of a particular FcR subtype.
Ligation and cross-linking of FcγRs initiates a cascade of biochemical signals that are initiated by activation of nonreceptor protein-tyrosine kinases of the Src family, which phosphorylate ITAM motifs in the FcγR chain and in FcγRIIa to recruit and activate the Syk tyrosine kinase and further amplify ITAM phosphorylation. Subsequent activation of several parallel pathways, including activation of PLC, PI3K, and MAP kinase (MAPK)-related pathways, and other targets leads to various cellular responses, including actin and membrane remodeling for particle ingestion, NADPH oxidase activation, and cytokine release. The DAP12 adaptor that normally potentiates these activating signaling pathways may signal for FcR and TLRs on macrophages that express TREM-2.
The opsonization of microbes with secretory IgA antibodies is likely to be important in the clearance of microbes from the mucosal surfaces of the respiratory, GI, and urogenital tracts. Neutrophils and monocytes have an FcR for IgA (FcαR) that has close structural similarities with other members of the FcR family. Ligation of the phagocyte FcαR can trigger phagocytosis, degranulation, and superoxide release. IgE antibodies can activate eosinophils and mast cells through the high-affinity FcεRI receptors.
Phagocytosis
Engagement of any of the opsonic receptors initiates phagocytosis of an opsonized particle to form a phagocytic vacuole that encloses the particle. The molecular details of this process are incompletely understood, but they involve membrane remodeling and the regulated assembly and disassembly of the actin cytoskeleton in a fashion analogous to the mechanisms that result in cell movement when chemotactic receptors are engaged. FcγR-mediated phagocytosis triggers the extension of long pseudopodia that attach in a zipperlike fashion around the particle and then fuse to form the phagosome, which is then drawn into the cell. In contrast, complement-opsonized particles attach to the cell surface only at limited points and “sink” into the cytoplasm in the absence of any pseudopod extension. These morphologic differences are associated with differences in the underlying biochemical pathways. FcR-mediated phagocytosis is sensitive to inhibitors of tyrosine kinases and requires the Rac and Cdc42 GTPases, whereas CR3-mediated phagocytosis is dependent upon protein kinase C and the Rho GTPase.
Microorganisms can also enter phagocytes by nonopsonic routes that enable them to evade the cytocidal weapons that are otherwise activated upon phagocytosis. For example, after binding to the macrophage cell surface, Salmonella typhimurium induces extensive membrane ruffling that leads to internalization of the bacterium into a macropinosome or “spacious phagosome.” Legionella pneumophila attachment to macrophages induces the formation of a pseudopod that spirals around the bacterium to form a “coiling phagosome” that does not acidify or fuse with lysosomes.
Cytocidal and Digestive Activity
The binding of ligands to phagocyte chemoattractant and opsonic receptors ultimately leads to the mobilization of phagocyte granules that contain cytotoxic and hydrolytic proteins and to the activation of enzymatic reactions that generate toxic oxygen metabolites ( Fig. 22-7 ). These complementary processes are designed to modify or destroy the inciting object and are often classified as oxygen-independent and oxygen-dependent pathways.
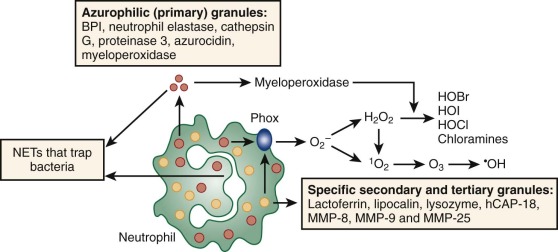
Neutrophil granules are secretory organelles that can be divided into four general classes, as discussed in “Neutrophil Granule Biosynthesis and Classification” (see Table 22-2 ). Degranulation (also referred to as exocytosis or mobilization ), the fusion of granule membranes with the plasma or phagosome membrane, results in the transfer of granule membrane constituents to a new membrane compartment and the discharge of the granule contents into the extracellular fluid or phagocytic vacuole. Microtubules and microfilaments are involved in granule translocation. Munc13-4, a regulator of vesicle trafficking, is essential for the recruitment of granule membranes to the surface and to the phagosomal membrane. Degranulation is triggered by increases in calcium concentration when neutrophils are activated through chemoattractant and other receptors. Different neutrophil granule populations have marked differences between expression of vesicle-associated membrane protein 2 (VAMP-2), a fusogenic protein involved in exocytosis, which is highest in secretory vesicles, less in gelatinase granules, and still lower in specific granules. Thus different granule classes vary in their responsiveness to calcium, which leads to the mobilization of different granule classes depending on the calcium concentration, which, in turn is proportional to the concentration of the chemoattractant or other activating signal. Secretory vesicles, whose membranes are storage pools for β 2 -integrin–adhesion proteins and other receptors, are mobilized with relatively low concentrations of calcium. The fusion of secretory vesicles with the plasma membranes provides a rapid way of upregulating the cell surface expression of these receptors along with MMP-25, another membrane constituent of secretory vesicles. Gelatinase (tertiary) granules are also easily mobilized for exocytosis at the cell surface to release the gelatinase (MMP-9). These metalloproteases are stored as inactive proforms and become activated by proteolysis after exocytosis to facilitate the breakdown of extracellular matrix during the early phases of neutrophil migration. At the opposite end of the spectrum, azurophil (primary) granules, which contain cytotoxic proteins and hydrolytic enzymes, undergo only limited exocytosis, fusing primarily with phagocytic vacuoles to deliver their contents into a sequestered compartment.
Monocytes and macrophages do not have populations of cytoplasmic storage granules equivalent to those found in neutrophils. Instead, internalized phagosomes fuse sequentially with endocytic vesicles, and subsequently lysosomes, to form a phagolysosome, which has a highly acidic interior rich in hydrolases.
Certain microorganisms can become intracellular parasites, because they have developed mechanisms to prevent granule fusion with the phagocytic vacuole or otherwise evade the phagocyte digestive and oxidative armamentarium. For example, although mycobacteria exist intracellularly within a phagosome, they produce compounds that inhibit their fusion with lysosomes. L. pneumophila and Toxoplasma may inhibit acidification and lysosomal fusion. Virulent strains of Salmonella engulfed by macrophages produce compounds that prevent translocation of the respiratory burst oxidase to the phagosomal membrane. Listeria monocytogenes escapes from the phagocytic vacuole altogether to avoid attack by lysosomal products and can survive in the cytoplasm of relatively quiescent macrophages and hepatocytes. Yersinia, group A streptococci, Helicobacter, Ehrlichia, and Francisella are examples of microbes that have developed similar strategies to survive in neutrophils.
In addition to delivering their antimicrobial granule contents to the interior of phagosomes, dying neutrophils are also capable of extruding weblike extracellular structures, NETs, composed of chromatin and granule proteins that can bind to microbes and may facilitate their killing. Formation of NETs appears to involve a novel process of cell death that in some cases is dependent on oxidants generated from the neutrophil NADPH oxidase. Since the discovery of this process, the molecular mechanisms of this process are emerging. It appears that intracellular liberation of elastase from azurophil granules, formation of hypochlorous acid, and decitrullination of histones to loosen the nucleosomes all play a role. Experimentally, several agents are used to induce NETs, phorbol myristate acetate being the most common. Not all stimuli induce NETs by the same mechanism and induction by bacteria or by soluble immune complexes are reported to be independent of MPO and the NADPH oxidase. Intracellular signals that lead to NETs, depending on the stimulus, include the Ref-MEK-ERK pathway, and protein kinase C isoforms, but also mammalian target of rapamycin–regulated expression of hypoxia-inducible factor 1α is involved in endotoxin-induced NET formation.
Neutrophils from newborns have a reduced capacity to form NETs and a correspondingly reduced capacity to kill bacteria extracellularly. This may relate to a reduction in the content of granule proteins including elastase in neutrophils from newborns.
The view of NETs as an alternative death pathway of neutrophils by which they still serve to defend against microbial invasion is now broadened by observations that demonstrate extrusion of DNA from neutrophils in circulation in a process that requires the participation of platelets to induce NET formation. Furthermore, neutrophils may extrude DNA to ensnare bacteria but maintain the capacity to migrate, albeit without the same clear orientation as before becoming anucleate. Formation of NETs are not only to the benefit of the host, because NETs have been shown to promote thrombosis and transfusion-related acute lung injury.
Oxygen-Independent Toxicity
Phagocyte granules supply preformed cytotoxic and digestive compounds that play a key role in oxygen-independent killing and digestion of microbes, senescent cells, and particulate debris. In neutrophils, azurophilic (primary) and specific (secondary) granules serve as the main storage reservoir of these compounds. Oxygen-independent pathways complement those dependent on the respiratory burst (see “Oxygen Dependent Toxicity”), and are also important for phagocyte antimicrobial activity under the adverse conditions of hypoxia and acidosis often encountered locally at the site of infection.
Numerous cationic antimicrobial proteins are contained within neutrophil azurophilic granules. Defensins are small (29-25 amino acids) basic peptides that constitute more than 5% of the total cellular protein of human neutrophils, although they are absent in murine neutrophils. These peptides exhibit antimicrobial effects against a broad range of gram-positive and gram-negative organisms, fungi, mycobacteria, and some enveloped viruses. Defensins are also cytotoxic to mammalian cells. Defensins kill target cells by insertion into the cellular membrane and formation of voltage-regulated channels. Defensinlike peptides have also been found in small intestinal Paneth cells and in tracheal epithelium. Bactericidal/permeability-increasing protein (BPI) is a 55-kD cationic protein that has potent cytotoxic effects toward gram-negative bacteria. BPI binds avidly to LPS, leading to both bacterial killing by damaging the cell membrane and to the neutralization of endotoxin associated with the bacterial cell wall and in serum. Serprocidins are a family of 29 kD glycoproteins that are homologous to members of the serine protease superfamily and include azurocidin (CAP37) and four serine proteases (cathepsin G, elastase, proteinase 3, and the recently identified NSP4). In human neutrophils, serprocidins are even more potent than the defensins in antimicrobial activity and have a broad spectrum of cytotoxicity that is, with few exceptions, unrelated to proteolytic activity. Cathepsin G, elastase, and proteinase 3 are often referred to as neutral proteases because the optimal pH for their proteolytic activity is approximately 7. Azurocidin can bind to endotoxin, which appears to account for its activity for gram-negative bacteria. Azurocidin also is a potent chemoattractant for monocytes as well as fibroblasts and T cells. Exogenous administration of antimicrobial peptides is being studied as an alternative or adjunctive therapy to conventional antibiotics in a number of settings.
Both azurophilic and specific granules contain lysozyme, which hydrolyzes the cell wall of saprophytic gram-positive organisms and may also assist in the nonlytic killing of other organisms. hCAP-18 is a member of the cathelcidin family of antimicrobial peptides, which is cleaved by proteinase-3 after exocytosis. The N-terminal region is homologous to other cathelicidins, whereas the 37-amino acid C-terminal fragment (LL-37) has additional activities, including acting as chemoattractant as well as effects on apoptosis. Specific granules also contain the iron-binding glycoprotein lactoferrin, which has direct bacteriocidal activity both related and unrelated to the chelation of iron compounds required for bacterial metabolism. Lactoferrin may also catalyze the nonenzymatic formation of hydroxyl radicals (OH•) during the respiratory burst (see “Oxygen-Dependent Toxicity”). Vitamin B 12 (cobalamin)–binding protein has been proposed to bind the analogous family of compounds found in bacteria to exert an antimicrobial effect. Lipocalin 2, also known as NGAL, interferes with bacterial iron utilization by binding to bacterial ferric-siderophore complexes. NGAL-deficient mice demonstrate a major reduction in defense against both Klebsiella pneumoniae and Mycobacterium tuberculosis, but NGAL, like most other neutrophil granule proteins, is highly expressed in epithelial cells during inflammation, and the role of neutrophil-delivered proteins versus epithelial-cell–fabricated proteins has not been determined. Human neutrophil NGAL has, however, been associated with defense against M. tuberculosis.
Azurophilic granules contain a variety of hydrolases (see Table 22-2 ) that have a lower pH optimum (<6), consistent with the lysosomal character of these granules. Studies using indicator dyes and biochemical techniques suggest that after a transient rise, the pH of the phagocytic vacuole falls below 6, which would enhance the activity of these enzymes upon their discharge into the vacuole. The acid hydrolases serve primarily a digestive rather than a microbicidal function. Azurophilic granules also contain MPO, which is an important enzyme in microbicidal oxygen-dependent reactions that are described in “Oxygen-Dependent Toxicity.”
Inherited partial or complete deficiency of MPO, which occurs in 0.05% of the population, can occasionally result in increased susceptibility to infection (see “Myelooxidase Deficiency”). Deficiencies in other individual neutrophil granule proteins have not yet been described in humans, but gene-targeted mice lacking the neutrophil granule serine proteases elastase or cathepsin G have impaired host defense against gram-negative sepsis and fungal infections. A few rare disorders involving defects in granule formation (SGD) or degranulation (CHS) are associated with recurrent bacterial infections. Inherited mutations in neutrophil elastase are present in patients with cyclic neutropenia (CyN) and severe congenital neutropenia (SCN). The mutant forms of elastase may have abnormal properties that exert a toxic effect on granulopoiesis, likely by eliciting an unfolded protein response that induces apoptosis.
Oxygen-Dependent Toxicity
The resting neutrophil relies primarily on glycolysis for energy and hence consumes relatively little oxygen. However, within seconds after contacting opsonized microbes or high concentrations of chemoattractants, oxygen consumption increases dramatically, often by more than 100-fold. This “extra respiration of phagocytosis” was first observed in 1933, but it was almost 30 years before it was appreciated that this process was insensitive to mitochondrial poisons and thus not related to increased energy demands. The enzyme complex responsible for this phenomenon, referred to as the NADPH or respiratory burst oxidase, is associated with the plasma and phagolysosomal membranes and catalyzes the transfer of an electron from NADPH to molecular oxygen, thereby forming the superoxide radical (O 2 −) ( Fig. 22-8 ). Superoxide, although itself a relatively weak microbicidal agent, is the precursor to a family of potent oxidants that are essential for the killing of many microorganisms (see Fig. 22-8 ). The importance of the respiratory burst to normal host defense is underscored by the recurrent and often life-threatening infections seen in patients with chronic granulomatous disease (CGD), who are genetically deficient in respiratory burst oxidase activity.
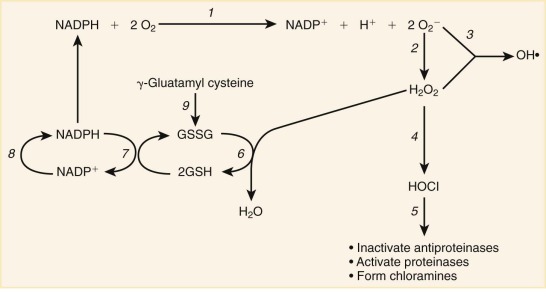
The respiratory burst oxidase is a multi-subunit enzyme complex assembled from membrane-bound and soluble proteins upon phagocyte activation ( Fig. 22-9 ). Five polypeptides, gp91 phox , p22 phox , p47 phox , p67 phox , and p40 phox , that are essential for normal respiratory burst function have been identified ( Table 22-7 ), and mutations in the corresponding genes are responsible for five different genetic subgroups of CGD. The oxidase subunits have been given the designation phox , for ph agocyte ox idase. Neutrophils have the highest expression level of the NADPH oxidase, followed by monocytes, macrophages, eosinophils, and dendritic cells. B lymphocytes also express the NAPDH oxidase, but at very low levels, and even smaller amounts are reported in T lymphocytes.
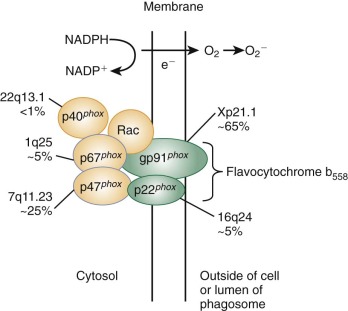
| gp91 phox (NOX2) | p22 phox | p47 phox | p67 phox | p40 phox | |
|---|---|---|---|---|---|
| Synonyms | β Chain Heavy chain | α Chain Light chain | NCF-1 SOC II | NCF-2 SOC III | NCF-4 |
| Amino acids | 570 | 195 | 390 | 526 | 339 |
| Gene locus | CYBB Xp21.1 | CYBA 16q24 | NCF1 7q11.23 | NCF2 1q25 | NCF4 22q.13.1 |
| Cellular location in resting neutrophil | Specific granule and secretory vesicle membranes Plasma membrane | Specific granule and secretory vesicle membranes Plasma membrane | Cytosol | Cytosol | Cytosol |
| Functional domains | Binding sites for heme and FAD NADPH binding sites for cytosolic oxidase components | Proline-rich domain in carboxy-terminal that binds p47 phox | 9 potential serine phosphorylation sites SH3 domains Proline-rich domains PX domain | SH3 domains Proline-rich domains TPR repeats that bind Rac-GTP PB1 domain | SH3 domain PX domain PB1 domain |
| Homologies | NOX protein family FNR Yeast ferric iron reductase | Polypeptide I of cytochrome c oxidase (weak homology) | NOXO1 Interacts with other NOXs | NOXA1 Interacts with other NOXs |
An unusual b-type flavocytochrome, located in the membrane of secretory vesicles, gelatinase granules, and specific granules, mediates electron transfer in the oxidase complex. It is often referred to as flavocytochrome b 558 , for its spectral peak of light absorbance at 558 nm, or as flavocytochrome b 245 in reference to its midpoint potential of −245 mV, which is extremely low. This flavocytochrome is a heterodimer that contains a 91-kD glycosylated protein, gp91 phox , and a nonglycosylated subunit, p22 phox . The gene for gp91 phox , which is the site of mutations in the X-linked form of CGD, was one of the first human disease–associated genes to be identified by positional cloning. gp91 phox is the redox center of the oxidase and contains both flavoprotein and heme-binding domains in its cytosolic and membrane-spanning portions, respectively. This subunit is also sometimes called NADPH oxidase (NOX) 2, referring to its number in a series of homologous flavocytochromes. The p22 phox subunit is also an integral membrane protein and provides an important docking site for p47 phox during NADPH oxidase assembly. Heterodimer formation with gp91 phox is essential for heme incorporation and intracellular stability of both flavocytochrome subunits.
The p47 phox , p67 phox , and p40 phox subunits are found in the cytosol as a complex that is stabilized by intermolecular interactions that include those mediated by SH3 domains and proline-rich SH3 binding motifs within these proteins. Phagocyte activation induces translocation of this complex to the flavocytochrome, which is triggered by the phosphorylation of p47 phox to expose additional SH3 domains that bind to a proline-rich target SH3-binding sequence in p22 phox . The p47 phox subunit is necessary for translocation of p67 phox to the membrane of activated neutrophils based on studies of p47 phox -deficient CGD neutrophils. However, substantial amounts of superoxide can be generated from neutrophil membranes in vitro in the absence of p47 phox , provided that high concentrations of p67 phox and Rac-GTP are supplied. Hence, p47 phox acts as an “adaptor” protein to mediate translocation of p67 phox and to position it correctly in the active NADPH oxidase complex. The p40 phox plays a specialized role in stimulating high levels of superoxide production on phagosome membranes via a domain that binds to the membrane lipid phosphatidylinositol 3-phosphate. p40 phox may also help with stabilization of p67 phox on flavocytochrome b–containing granule membranes as they are recruited to the phagosomes.
The active NADPH oxidase also requires the GTP-bound form of the Rho GTPase, Rac. The main target of Rac-GTP is the p67 phox subunit, which contains a Rac-binding domain created by four α-helical tetratricopeptide repeat motifs. The GTP-bound form of Rac also appears to interact with flavocytochrome b .
Oxidase assembly is triggered by receptor-mediated binding of many soluble chemoattractants (see Table 22-4 ), which requires higher concentrations of these molecules compared with the initiation of chemotaxis. The binding of opsonized microbes to Fcγ and complement receptors or of fungi to β-glucan receptors are other major physiologic triggers of the respiratory burst, which is activated at sites of contact. Two critical events downstream of receptor binding are the phosphorylation of p47 phox and the activation of Rac to its GTP-bound state. The functional oxidase complex is assembled at the plasma membrane in response to soluble agonists. During phagocytosis, additional flavocytochrome b for NADPH oxidase assembly is delivered to the phagosome by the membrane fusion of specific granules, which contain the majority of the neutrophil’s supply of flavocytochrome b . Since release of O 2 − occurs largely at the extracellular side of the membrane, oxidants are released at sites of microbial contact or within the phagocytic vacuole, where they can interact with granule contents to potentiate their microbicidal effects.
Based on the redox properties of the flavocytochrome b 558 , the following pathway has been proposed for transfer of electrons from NADPH to oxygen (O 2 ) in the respiratory burst (see reaction 1 in Fig. 22-8 ):
| NADPH | flavin | heme | O 2 | O 2 − |
| → | → | → | → | |
| −330 mV | −256 mV | −245 mV | −160 mV |
The flavocytochrome spans membrane, so that NADPH is oxidized at the cytoplasmic surface and oxygen is reduced to form O 2 − on the outer surface of the plasma membrane (or inner surface of the phagosomal membrane).
Once formed, the O 2 − radical is first converted, either spontaneously or by means of superoxide dismutase, into hydrogen peroxide (H 2 O 2 ; see reaction 2 in Fig. 22-8 ). Azurophilic granule (derived MPO) in the presence of halides catalyzes the conversion of H 2 O 2 to hypochlorous acid (HOCl), the active agent in household bleach (see reaction 4 in Fig. 22-8 ). H 2 O 2 may also be converted into OH• in a nonenzymatic reaction with O 2 − catalyzed by either iron or copper ions (see reaction 3 in Fig. 22-8 ). H 2 O 2 , HOCl, and OH• are all strong oxidants that participate in microbial killing within the phagocytic vacuole. Reactive oxidants also regulate phagocyte proteolytic activity by activating latent phagocyte metalloproteinases (such as collagenase and gelatinase) and inactivating plasma antiproteinases. Enhanced phagocyte proteolysis at localized sites may be important for facilitating cellular migration into inflamed tissues, destruction of microbes, and removal of cellular debris.
Other enzymatic pathways related to oxidant generation include the detoxification of H 2 O 2 by glutathione peroxidase and reductase (see reactions 6 and 7, Fig. 22-8 ). Glutathione is produced from γ-glutamyl cysteine by the enzyme glutathione synthetase (see reaction 9, Fig. 22-8 ). Other important antioxidant systems in phagocytes and other tissues include catalase, which catalyzes the conversion of H 2 O 2 into O 2 and water; ascorbic acid; and α tocopherol (Vitamin E). The generation of NADPH is important in providing a source of reducing equivalents for the glutathione detoxification pathway as well as the respiratory burst itself. NADPH is replenished from NADP + by leukocyte glucose-6-phosphate dehydrogenase (G6PD; see reaction 8, Fig. 22-8 ) in the hexose monophosphate shunt.
A second oxygen-dependent pathway with antimicrobial effects involves the generation of NO from the oxidation of L-arginine to L-citrulline. This reaction is catalyzed by NO synthase (NOS), with molecular oxygen supplying the oxygen in NO. There are three different NOSs, two of which are constitutively expressed in a variety of tissues, including endothelium, brain, and neutrophils. Expression of a third, high-output isoform of NOS (inducible NOS [iNOS]) is inducible by inflammatory stimuli in a variety of cells, including macrophages and neutrophils, where it has a wide spectrum of antitumor and antimicrobial activity against bacteria, parasites, helminths, viruses, and tumor cells. NO can also interact with superoxide to form peroxynitrite, which mediates tyrosine nitration of cellular and bacterial proteins. High levels of iNOS-catalyzed NO production are readily elicited in normal mouse macrophages by exposure, for example, to IFN-γ and endotoxin. In human monocytes and macrophages, IFN-α/β, IL-4 plus anti-CD23, or chemokines can induce iNOS expression. The expression of iNOS has been detected in a variety of inflammatory and infectious diseases in humans, including malaria, hepatitis C, tuberculosis, tuberculoid leprosy, and acquired immunodeficiency syndrome dementia. Cytokine-activated human neutrophils also exhibit inducible production of NO that leads to nitration of ingested bacteria.
Specialized Functions of Mononuclear Phagocytes
Mononuclear phagocytes, particularly tissue macrophages, participate in a broad range of activities important for tissue homeostasis and repair as well as in the host defense against viruses, bacteria, fungi, and protozoa ( Box 22-1 ). From the standpoint of antimicrobial function, activated macrophages play a key role in the ingestion and killing of intracellular parasites, such as mycobacteria, Listeria, Leishmania, Toxoplasma, and some fungi, although some intracellular organisms have evolved specialized evasion mechanisms. Both oxygen-dependent and oxygen-independent systems are involved in this process, as described. When microbes (most characteristically mycobacteria, but any other organism or particle) cannot be fully ingested or killed, macrophages fuse in a cytokine-mediated process to form giant cells and granulomas.
Inflammatory Response and Pathogen Control
Antimicrobial activity
Antiviral activity
Antitumor activity
Secretion of cytokines, eicosanoids, proteases, coagulation factors
Granuloma formation
Immune Regulation
Antigen processing and presentation
Secretion of cytokines and chemokines
Tissue Homeostasis and Repair
Scavenger function
Removal of apoptotic, senescent, or necrotic cells
Phagocytosis of debris
Wound repair
Débridement and phagocytosis
Secretion of growth factors for endothelial cells and fibroblasts
Hematopoiesis
Secretion of growth factors
Iron metabolism
Lipid metabolism
IFN-γ, one of the principal macrophage-activating factors, is secreted by T lymphocytes as well as by macrophages and neutrophils. The cytokine induces changes in macrophage gene expression through the JAK–signal transducer and activator of transcription (STAT) pathway, elements of which are shared by many other cytokines, including IL-2, IL-6, and G-CSF. Endotoxin, the bacterial LPS derived from gram-negative bacteria, is another important trigger of macrophage activation through pathways involving CD14 and TLRs.
The classically activated, or M1 macrophages, are induced and supported by IFN-γ or IFN-β in combination with TNF-α. NK cells or other IFN-γ-producing lymphocytes can initiate this activation, and it may be sustained by an autocrine loop where INF-β and TNF-α are induced by danger signals sensed through TLRs expressed on the macrophage. M1 macrophages are indispensable for killing of intracellular pathogens and are also producers of proinflammatory cytokines such as TNF-α, IL-1, IL-6, Il-12, and IL-23. These are also potent activators of T-helper cells (Th1 and Th17). Macrophages may also develop into the alternatively activated or M2 phenotype that have antiinflammatory functions and promote tissue healing. IL-4 and IL-13 are key inducers of M2 macrophages. These cytokines are primarily produced by eosinophil and basophils and CD4+ type 2 helper T cells (Th2 cells); also IL-10, IL-21, and GM-CSF support the M2 phenotype. The M2 macrophages secrete antiinflammatory cytokines, primarily IL-10 and TGF-β. These macrophages are important for resolution of inflammation.
A particular activity of macrophages is the inhibition of antitumor activity of T cells. This is ascribed to myeloid-derived suppressor cells (MDSCs) that share many functional activities with M2 macrophages. Neutrophil subsets have also been described that function as inhibitors of T-cell function. A common feature of these MDSCs is the ability to deplete T cells of arginase. The development of MDSCs is supported by local factors secreted by tumors, such as TGF-β and GM-CSF. Because many tumors induce a systemic inflammatory response, at least neutrophil MDSCs may represent immature neutrophils liberated from the bone marrow. LPS induces liberation of such immature neutrophils with features characteristic of MDSCs in blood.
Interactions of macrophages with T and B cells are essential for the development of cellular and humoral immunity. Macrophage production of IL-1, and subsequent interactions between stimulated T and B cells, leads to B cell production of antigen-specific immunoglobulins. Macrophages are also major physiological sources of cytokines and chemokines that regulate both the innate and adaptive immune systems. As a source of “endogenous pyrogens,” they are responsible for the production of fever in response to infection or inflammation.
Macrophages participate in many aspects of wound repair. The early phases of this process are dominated by an influx of neutrophils, followed by the migration of monocytes that differentiate into activated macrophages, and finally, the appearance of T lymphocytes. Proliferating fibroblasts secrete collage and other matrix proteins important for wound closure and tissue remodeling, and migrating keratinocytes regenerate the epithelial surface. Both neutrophils and macrophages protect against infection and dispose of phagocytosed debris. Mononuclear phagocytes also elaborate fibroblast, epithelial, and angiogenic growth factors ( Box 22-2 ), which stimulate the normal progression of tissue repair and neovascularization that characterize the later phases of wound healing.
|
|
Macrophages ingest and dispose of apoptotic and necrotic cells. This function not only contributes to cellular processing for antigen presentation but also plays a critical role in tissue remodeling and homeostasis, embryological development, and the resolution of inflammation. A redundant and promiscuous system of receptors including integrins, scavenger receptors, complement receptors, calreticulin, CD14, and Mer receptor recognize and effect the uptake of apoptotic cells. Resident tissue macrophages, particularly those lining the sinusoids of the spleen and liver (formerly known as the reticuloendothelial system ), clear senescent and antibody-bound blood cells from the circulation.
Iron from the catabolism of hemoglobin in aged erythrocytes is incorporated into ferritin and hemosiderin, where it accounts for about two-thirds of the body’s store of reserve iron. Iron in this macrophage storage pool turns over and returns in a transferrin-bound form to the bone marrow for new red blood cell synthesis (see Chapter 11 ). Sequestration of iron in macrophages leads to the anemia of chronic disease. This is largely regulated by hepcidin, a small peptide synthesized by the liver in response to Il-6, which binds to and induces degradation of ferroportin, the mediator of iron export from macrophages and enterocytes.
Monocytes and macrophages contribute to the pathophysiology of atherosclerosis through the uptake, metabolism, and oxidation of LDL and very low density lipoprotein by receptor-mediated endocytosis. Macrophages in blood vessel walls that are exposed to sufficient quantities of cholesterol develop into foam cells characteristic of atherosclerotic plaques and contribute to the important inflammatory component of plaque formation and destabilization.
Specialized Functions of Eosinophils and Basophils
Eosinophils and basophils, which share many of the functional characteristics of neutrophils and mononuclear phagocytes, participate in distinctive aspects of the inflammatory response and interact with each other in the context of certain allergic reactions. Eosinophils and mast cells are often situated beneath epithelial surfaces exposed to environmental antigens, such as the respiratory and GI tracts, where they may be actively involved in mucosal immune responses. However, the role of eosinophils, basophils, and mast cells are better known in pathologic settings than in normal homeostasis.
Eosinophils appear to have both immunoenhancing and immunosuppressive functions ( Table 22-8 ). Although capable of ingesting and killing bacteria, eosinophils are not particularly efficient at this task. Rather, they possess an unusual ability to destroy invasive metazoan parasites, especially helminthic parasites. Eosinophils bind to the surface of both adult and larval helminths and inflict damage through release of cationic granule proteins and by the generation of reactive oxidants, including the eosinophil peroxidase–catalyzed formation of hypohalous acids via the action of the respiratory burst and eosinophil peroxidase.
| Function | Mechanism |
|---|---|
| Defense against helminths (both larval and adult forms) | Binding of eosinophils to surface Peroxidation of larval surface mediated by eosinophil peroxidase Toxicity to larval surface by released major basic protein (MBP) |
| Immunosuppression of immediate hypersensitivity reactions | Engulfment of most cell granules Release of prostaglandin E 1 /E 2 to suppress basophil degranulation Release of histaminase Oxidation of slow-reacting substance of anaphylaxis Release of phospholipase D to inactive mast cell platelet-activating factor Release of MBP for binding of mast cell heparin Release of plasminogen to reduce local thrombus formation |
Eosinophil production of the lipid inflammatory mediators, leukotriene C4 and PAF, play a role in the pathogenesis of allergic diseases. PAF and leukotriene C 4 can induce smooth muscle contraction and promote the secretion of mucous, and PAF itself is a potent activator of eosinophils. The release of eosinophil granule contents may also contribute to localized tissue damage. Purified eosinophil MBP, for example, can cause cytopathic changes in tracheal epithelium in vitro that are similar to the changes observed in asthmatic patients.
Eosinophils may also perform an immunosuppressive function in immediate hypersensitivity reactions (see Table 22-8 ). IgE-activated basophils or mast cells release eosinophilic chemotactic factor of anaphylaxis, which recruit eosinophils to the site. Subsequent eosinophil degranulation releases products that can inactivate inflammatory mediators. For example, histaminase inactivates histamine, phospholipase B inactivates PAF, MBP inactivates mast cell heparin, and lysophospholipase prevents the generation of arachidonic acid metabolites.
Basophils and mast cells are central participants in a variety of inflammatory and immunologic disorders, particularly immediate hypersensitivity diseases, and they may also play a role in host defense against bacterial infections. Basophils and mast cells express plasma membrane receptors that specifically bind with high affinity the Fc portion of the IgE antibody (Fcε receptors). After active or passive sensitization with IgE, exposure to specific multivalent antigen triggers an almost immediate release of granule contents (anaphylactic degranulation) and the synthesis and release of newly generated chemical mediators such as leukotriene C 4 , which stimulates smooth muscle contraction, mucous secretion, and vasoactive changes. Degranulation can also be triggered in response to insect venoms, radiocontrast dye, and other irritants. Studies conducted in 1994 on mutant mice engineered by gene targeting to lack Fcγ and/or Fcε receptors have shown that mast cell FcγRIII receptors are essential in activating the inflammatory response to IgG immune complexes (Arthus reaction), heretofore an unrecognized role for the mast cell. Mast cells can secrete numerous mitogenic or inflammatory cytokines, including many ILs (1, 3, 4, 5, and 6), chemokines, GM-CSF, and TNF-α, that are also likely to regulate leukocyte recruitment and inflammation in IgE-dependent reactions and immune complex injury. Inflammatory cytokines and leukotrienes released from tissue mast cells have been recognized to play an important role in neutrophil recruitment during the acute response to bacterial infection.
Pathologic Consequences of Phagocyte Activation and Inflammatory Response
Although it normally serves a protective function, the inflammatory response can also result in damage to host tissues. The release of proteases, oxygen radicals, and proinflammatory cytokines by activated phagocytes appears to play a major role in the generation of tissue injury in a wide variety of pathologic inflammatory processes ( Box 22-3 ). For example, neutrophil elastase has been implicated in the pathogenesis of emphysema in both adult smokers and individuals with α1-antitrypsin deficiency. Neutrophil granule proteases may contribute to the joint destruction in rheumatoid arthritis and other chronic arthropathies. Neutrophils are also believed to play a key role in the systemic inflammatory response syndrome, a term that has been created to encompass the host response to both infectious (e.g. gram-negative sepsis) and noninfectious (e.g. pancreatitis, trauma) etiologies, and can lead to organ dysfunction and tissue damage. Sequestration of activated neutrophils in the pulmonary capillary bed and subsequent release of tissue-damaging agents is an important component in the development of adult respiratory distress syndrome. Activation of the complement cascade by artificial membrane surfaces during hemodialysis and cardiopulmonary bypass also can result in neutrophil activation, intrapulmonary sequestration, and lung injury. Macrophages are integral to the pathophysiology of atherosclerosis by uptake of serum lipoproteins and contribute an important inflammatory component that influences atherosclerotic plaque development and rupture. In addition to their cytotoxic effects, oxidative products released by activated phagocytes are also mutagenic, as documented by plasmid mutagenesis, sister chromatid exchange, and transformation of cells in culture. Hence the increased risk of malignancy observed with certain chronic inflammatory states, such as ulcerative colitis or chronic hepatitis, has been postulated to be in part related to oxidant-induced carcinogenesis.
Arthus reaction
Systemic inflammatory response syndrome
Nephrotoxic and immune complex nephritis
Postischemic myocardial damage
Adult respiratory distress syndrome
Atherosclerosis
Bronchiectasis
Acute and chronic allograft rejection
Malignant transformation with chronic inflammation
Rheumatoid arthritis
The development of antiinflammatory interventions based on agents that block leukocyte adhesion or inhibit the action of specific proinflammatory agents has been an area of intense interest. IL-1 receptor antagonists are used to treat autoinflammatory diseases such as familial Mediterranean fever as well as rheumatoid arthritis and are being evaluated in a broad range of inflammation-associated states. TNF-α antagonists are now widely used for treatment of inflammatory bowel disease, rheumatoid arthritis, and other autoimmune disorders. Antagonists of leukocyte integrins have shown benefit in phase II and III studies for inflammatory bowel disease, psoriasis, and multiple sclerosis. However, although protective effects of monoclonal antibodies directed against β 2 integrins, ICAM-1, or selectins were found in various animal models of inflammation including ischemia-reperfusion injury, endotoxic shock, and acute arthritis, results from clinical trials failed to show significant benefit. The contributions of nonphagocytic cells to inflammatory tissue injury must also be kept in mind. For example, the adult respiratory distress syndrome can occur in the presence of severe neutropenia. Moreover, despite the adverse consequences of the acute inflammatory process, these events are also important for normal healing. For example, the use of antiinflammatory agents in myocardial infarction, which can decrease infarct size acutely, results in impaired healing of the myocardium and the formation of fragile scar tissue. Finally, the impact of the new antiinflammatory biologic agents on host defense must be kept in mind, particularly if such agents are being used in combination with other immunosuppressive drugs such as steroids. For example, patients receiving TNF-α antagonists have increased rates of tuberculosis and infections with endemic mycosis or intracellular bacterial pathogens.
Quantitative Granulocyte and Mononuclear Phagocyte Disorders
This section reviews clinical disorders associated with disturbances in granulocyte number, with particular emphasis on syndromes in which the granulocyte abnormality is a central feature. Several disorders have now been associated with specific genetic defects, although the molecular pathophysiology leading from the gene to the phenotype often remains obscure. This section of the chapter is organized by clinical characteristics of the disorders, with genetic and functional laboratory data provided where available.
Neutropenia
Definition and Classification
Neutropenia is defined as a decrease in the absolute number of circulating neutrophils in the blood. Normal neutrophil levels should be stratified for age, race, and other factors. For whites, the lower limit for normal absolute neutrophil count (ANC; total neutrophils and bands) is 1000 cells per µL in infants between 2 weeks and 1 year of age; then it rises after infancy to 1500 cells per µL. Persons of African descent have somewhat lower neutrophil counts, with ANC less than 1500 cells per µL in 4.5% of African-American participants in one U.S. survey. This is entirely attributable to the lower ANC in the subpopulation with the Duffy negative (Fy−/−) blood group. This blood group, selectively enriched in populations in the malarial belt of Africa, is associated with ANCs 200 to 600 cells per µL less than those who are Duffy positive. Falsely low white blood cell counts can result when counts are done long after blood is drawn or after leukocyte clumping in small clots or in the presence of certain paraproteins.
Neutropenia can represent disturbances in neutrophil production, altered distribution of neutrophils between the circulating and the marginated or tissue pools (pseudoneutropenia), increased peripheral utilization or destruction, or combinations of these causes. However, assays of leukokinetics and myelopoiesis are not routinely available, so mechanisms may be hard to identify. Classifications based on biochemical or functional studies are also difficult because of the paucity of neutrophils in the circulation of neutropenic patients. In general, neutropenic syndromes are broadly classified as intrinsic because of defects in myelopoiesis or as acquired, as a result of extrinsic factors such as drugs, infections, or autoantibodies ( Box 22-4 ). Congenital neutropenias are now classified largely on the basis of known genetic defects.
Neutropenia Caused by Intrinsic Defects in Granulocytes or Their Progenitors *
* Types of neutropenia are listed in order of discussion in the text.
Reticular dysgenesis
Severe congenital neutropenia
Cyclic neutropenia
Myelokathexis/WHIM syndrome
Shwachman-Diamond syndrome
Albinism/neutropenia syndromes (including Chédiak-Higashi syndrome)
Familial benign neutropenia
Bone marrow failure syndromes (congenital and acquired)
Neutropenia associated with immune deficiency disorders
Neutropenia associated with metabolic disorders
Neutropenia Caused by Extrinsic Factors
Infection
Drug-induced
Autoimmune
Neonatal immune
Metabolic diseases
Nutritional deficiencies
Reticuloendothelial sequestration
Bone marrow infiltration
Chronic idiopathic neutropenia (may also be intrinsic)
WHIM, Warts, hypogammaglobulinemia, infections, myelokathexis.
The clinical significance of neutropenia depends upon the level of depression of the ANC. Mild neutropenia with neutrophil counts of 1000 to 1500 cells per µL and moderate neutropenia with counts of 500 to 1000 cells per µL are generally not clinically significant with respect to host defense, but these situations may warrant evaluation to determine the underlying cause. Severe neutropenia refers to an ANC below 500 cells per µL, with the term agranulocytosis usually reserved for counts below 200 cells per µL. Children with severe chronic neutropenia can be registered with the Severe Chronic Neutropenia International Registry (SCNIR; http://depts.washington.edu/registry/ ).
Symptoms of Neutropenia
The hallmark of neutropenia is an increased susceptibility to bacterial and fungal infection; neutropenia by itself does not diminish host defense to viral or parasitic infections. The most common types of pyogenic infections in patients with significant neutropenia are cellulitis, furunculosis, superficial or deep cutaneous abscesses, pneumonia, and septicemia. Stomatitis, gingivitis, and periodontitis may be presenting signs of neutropenia that develop into chronic problems. Perirectal inflammation and otitis media (especially in younger children) occur as well. Endogenous bacteria are the most common cause of infections, but colonization or infection may occur with a variety of nosocomial and opportunistic organisms. The most commonly isolated organisms from neutropenic patients are Staphylococcus aureus and gram-negative bacteria, but almost any organism can appear, particularly in hospitalized patients or those with frequent or prolonged courses of antibiotics. The usual symptoms and signs of local infection—such as exudates, fluctuation, ulceration, and regional adenopathy—may be less evident in patients with neutropenia than in normal individuals.
Susceptibility to bacterial infection, even with severe neutropenia, can be quite variable, depending on the underlying etiology. For example, some patients with chronic neutropenia resulting from autoantibodies do not experience serious infections over a period of many years even with neutrophil counts as low as 200 cells per µL, most likely because these individuals have rapid production and transit of normal neutrophils with relative preservation of neutrophil delivery to tissues. Many patients with chronic neutropenia also have normal to increased numbers of circulating monocytes, an alternative phagocyte. However, the recruitment of monocytes to inflammatory sites is delayed relative to neutrophils, and monocytes are not as efficient as neutrophils in ingesting bacteria. Thus monocytes appear to provide only marginal protection against pyogenic organisms in patients with severe neutropenia. It is likely that the humoral, cell-mediated, and tissue macrophage immune systems also play important alternative or compensatory roles preventing infection in these individuals.
Neutropenia Caused by Intrinsic Defects in Granulocytes or Their Progenitors
These disorders are also discussed in Chapter 8 .
Reticular Dysgenesis.
The selective failure of stem cells committed to myeloid and lymphoid development leads to reticular dysgenesis, one of the rarest and most severe forms of severe combined immunodeficiency. It is characterized by severe leucopenia—including both agranulocytosis and lymphopenia; defective cellular and humoral immunity functions; and absence of lymph nodes, tonsils, Peyer’s patches, and splenic follicles—as well as sensorineural hearing loss. Erythroid and megakaryocyte development is normal. All reported infants have died of bacterial or viral infections within the first weeks of life, except a few treated with hematopoietic stem cell transplantation (HSCT). The underlying genetic defects in this AR disorder are biallelic mutations in the AK2 gene, encoding the mitochondrial energy metabolism enzyme adenylate kinase 2. As discussed below, other mitochondrial enzyme deficiencies have been associated with isolated neutropenia.
Severe Congenital Neutropenia.
SCN was first described by Kostmann in 1956 as an AR disorder associated with severe neutropenia that was identified in the population of an isolated northern parish of Sweden. Other forms of SCN have since been identified with de novo occurrence and AD, AR, or X-linked inheritance. SCN (Online Mendelian Inheritance in Man [OMIM] # 202700) refers to the entire disorder regardless of inheritance or genotype, whereas Kostmann disease (OMIM # 610738) refers to the AR subtype caused by mutations in the HAX1 gene (see later in this section). This disorder is also discussed in Chapter 8 .
The incidence of SCN is approximately two cases per million population. Affected patients generally maintain ANCs below 200 cells per µL, which has been documented on the first day of life in several cases. Episodes of fever, skin infections (including omphalitis), stomatitis, pneumonia, and perirectal abscesses typically appear during the first months of life. Infections often disseminate to the blood, meninges, and peritoneum, and are usually caused by S. aureus , Escherichia coli, and Pseudomonas species. Before the current era of G-CSF therapy, most patients died in the first 1 to 2 years of life, and survivors were known to be at risk for developing myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML).
Peripheral blood counts generally show, in addition to agranulocytosis, moderate to marked monocytosis, mild to moderate eosinophilia, and secondary changes such as anemia and thrombocytosis. G-CSF production in patients with SCN appears to be normal, if not elevated. Antineutrophil antibodies may be present, and a positive test should not be used to rule out SCN.
Bone marrow examination characteristically shows a myeloid “maturation arrest” at the myelocyte stage of development, with normal to increased numbers of promyelocytes but few if any more mature forms. The promyelocytes may show dysplastic morphology, including large size, atypical nuclei, and vacuolated cytoplasm. Marrow eosinophilia and monocytosis is common and does not change during treatment. Cellularity is usually normal or slightly decreased. Megakaryocytes are normal in number and morphology.
The genetic basis of 40% to 80% of SCN cases derives from mutations in the ELANE gene (formerly termed ELA2 ), which encodes neutrophil elastase and is also responsible for the related but distinct disorder, CyN (discussed later in this section). ELANE -related SCN may arise sporadically by de novo mutation or appear in kindreds with an AD Mendelian inheritance pattern, most dramatically illustrated by the six affected children from five families, all conceived by sperm from the same anonymous donor.
The SCNIR analyzed 188 patients with SCN who had 94 mutations and 119 patients with CyN who had 22 mutations (see Fig. 22-10 ). Most ELANE mutations resulted in only an SCN or the cyclic phenotype ( Fig. 22-10 ), but 12 mutations were observed in both patients with CyN and SCN. A number of recurring mutations were observed only in CyN, including Q97L, D174ins, and W241G and conversely, a number of recurring mutations were observed only in SCN, including A57T, R103P, and G214R.
How mutations in neutrophil elastase result in the cyclical production of neutrophils and other blood cells is not entirely understood. Bone marrow samples from both patients with CyN and patients with SCN demonstrate increased apoptosis of neutrophil precursors. Thus expression of mutant neutrophil elastase appears to induce excessive apoptosis, most likely through either aberrant subcellular targeting of the protein or induction of a strong unfolded protein response.
Additional very rare cases of AD SCN appear to arise from mutations in the Gfi1 gene, which encodes a transcriptional repressor that inhibits multiple genes involved in neutrophil maturation, including ELANE . Resultant overexpression of ELANE could lead to apoptosis by either of the mechanisms discussed previously.
AR SCN can be caused by defects in several genes, including HAX1 , an antiapoptotic gene that is responsible for the classic Kostmann syndrome, and G6PC3 . HAX1 encodes a multifunctional protein that is involved in the regulation of apoptosis, as well as cell motility, signal transduction, and mRNA processing. Other genes associated with AR SCN include VPS45 , which encodes an endosomal trafficking regulator, and G6PC3 , which encodes a neutrophil-specific catalytic subunit of glucose-6-phosphatase and may thus be considered functionally similar to glycogen storage disease type 1b (see later discussion in this section) but with pathology limited to the neutrophil. In contrast to ELANE , which is expressed only in myeloid cells, these genes are broadly expressed in many tissues and organs. As a result, some patients with HAX1 protein defects have associated neurological deficits; G6PC3 defects were originally described in a cohort of patients with cardiac anomalies, urogenital abnormalities, and venous angiectasias ; and VPS45 mutations are associated with neutrophil dysfunction, bone marrow fibrosis, and nephromegaly. However, both HAX1 and G6PC3 mutations can also be responsible for isolated neutropenia without an extended phenotype. G6PC3 has also been associated with rare cases of atypical CyN, indicating that, as for ELANE , the same mutation can produce either the severe or CyN phenotype depending upon the genetic background.
Other rare mutations responsible for an SCN phenotype include gain-of-function mutations in the X-linked Wiskott-Aldrich syndrome (WAS) gene and constitutional defects (as opposed to acquired mutations; see later) in the gene encoding the G-CSF receptor. Overall, approximately 60% of patients with a diagnosis of SCN can now be given a precise genetic diagnosis.
G-CSF, the standard treatment modality for SCN, has greatly improved both life span and quality of life for these patients. Until the availability of this cytokine in the 1980s, severe neutropenia would inevitably lead to fatal infections in the majority of patients, despite supportive care with prophylactic trimethoprim-sulfamethoxazole, aggressive use of antibiotics at the time of documented infections, and scrupulous attention to oral hygiene. More than 95% of patients with SCN respond to G-CSF treatment with an increase in the ANC to greater than 1000 per µL, with documented reduction in infections. There is often a delay of 7 to 10 days before the rise of peripheral blood neutrophil counts at the start of treatment. In patients enrolled in the SCNIR between 1993 and 1999, more than 90% of patients with congenital neutropenia responded to G-CSF treatment with rises in ANC to more than 1 ×10 9 /l and required significantly less antibiotic therapy and fewer hospitalizations. SCN patients required G-CSF doses ranging from 0.01 to 242 µg/kg/day (mean 12.6 µg/kg/day and standard deviation [SD] 19.3 µg/kg/day). These doses are significantly higher than those needed for cyclic or idiopathic neutropenia. The percentage of maturing neutrophils in the bone marrow increases with G-CSF, although morphologically abnormal promyelocytes with vacuolized and asynchronous nuclear-cytoplasmic maturation can persist, and the number of monocytes and eosinophils in the bone marrow and peripheral blood may remain high.
Based on data from the SCNIR, a starting daily dose of G-CSF at 5 µg/kg/day is recommended, increasing gradually if necessary to as high as 100 µg/kg/day for at least 14 days at each dose to achieve a neutrophil count of 1000 to 2000 per µL. The drug is generally administered subcutaneously but may be given at equal dosage intravenously if clinical conditions warrant or if vehicle volumes are too high to permit comfortable subcutaneous injection. In G-CSF refractory patients, combination therapy with SCF has been reported to be effective but rarely used, because it also leads to mast cell activation with histamine release and anaphylactoid reactions. GM-CSF treatment results in large increases in eosinophils and monocytes, but not neutrophils.
Side effects of G-CSF treatment are usually (but not always) mild and include bone pain, headache, and rashes. Other common but often asymptomatic findings include splenomegaly and osteopenia. Splenomegaly has been reported in patients with SCN before and with G-CSF therapy. Palpable splenomegaly, with a median spleen measurement of 2 cm below costal margin was recorded in 18.2% of untreated patients registered in the SCNIR. During the first year of G-CSF therapy, the incidence increased to 38.2% and remained at this level of occurrence (26.7% to 44.7%) through 10 years of therapy, with no trend to progression in spleen size on therapy.
Of 128 SCNIR subjects analyzed in 2010, including 40 children younger than age 21 years and 122 subjects receiving G-CSF, 46% of adults met criteria for osteopenia (t score −1), 9% met criteria for osteoporosis (t score <−2.5), and17.5% of children had low bone mineral density for age (Z score <−2.0). In children but not adults, bone mineral density was inversely related to duration of G-CSF therapy. The SCNIR recommends regular monitoring of bone density. Rare patients have also been found to have vasculitis or glomerulonephritis, both before and during G-CSF therapy.
The SCNIR has reported thrombocytopenia (platelet count <50 × 10 3 /µL) in 26 patients, including 17 with SCN. Among the 19 patients receiving G-CSF, seven developed thrombocytopenia concurrent with conversion to MDS/AML. Thus development of thrombocytopenia in SCN patients requires thorough evaluation as a possible heralding sign of disease conversion.
Conversion to MDS/AML and bacterial sepsis remain the most serious complications of SCN. The predilection to acute leukemia was recognized before the availability of G-CSF, when most affected children died early in life from severe infections. Now longer survival and G-CSF itself combine to produce, after 10 years of therapy, an ongoing 2.3% per year risk of developing AML or MDS that almost invariably progresses to AML.
During long-term therapy with G-CSF, some SCN patients acquire mutations in the G-CSF receptor gene ( CSF3R ), particularly C-terminal truncation mutations that confer a clonal growth advantage. Acquisition of these mutations is often, but not always, followed by evolution to myelodysplasia characterized by monosomy 7 and finally myeloid leukemia. Acquired mutations in CSF3R have been detected in approximately 80% of SCNIR patients who developed MDS/AML, whereas mutations in the FLT3, KIT, and JAK2 tyrosine kinase genes commonly found in de novo AML are uncommon. RUNX1 mutations appear most often upon evolution to AML, but genome-wide analysis has revealed acquired mutations in multiple genes, including CSF3R, LLGL2, and ZC3H18 in a subpopulation of progenitor cells early in the process, followed by mutations in known AML-associated genes ( RUNX1 and ASXL1 ) and chromatin remodelers ( SUZ12 and EP300 ) in leukemic clones.
Some patients with SCN and CSF3R mutations have not progressed, and there are some who lost the mutated clone upon discontinuation of G-CSF therapy and reacquired it upon resumption of treatment. Cytogenetic abnormalities, particularly monosomy 7, also precede and predict leukemic conversion. For this reason, yearly bone marrow surveillance, including karyotyping and chromosome 7 fluorescence in situ hybridization (FISH), should be performed on all patients with SCN, whether or not they are treated with G-CSF. Although clonal cytogenetic abnormalities may spontaneously remit, their appearance should be considered a strong indication for HSCT, including use of a matched unrelated donor if necessary, because transplantation is more likely to be successful when undertaken before progression to MDS/AML.
Recent studies by the SCNIR and the French Severe Chronic Neutropenia Study Group have shed light on the incidence and some of the risk factors for sepsis and MDS/AML. Although early reports suggested a rising risk of MDS/AML after 10 years of G-CSF therapy, longer follow-up study has shown the risk to be stable at 2.3% per year after 10 years of G-CSF therapy ( Fig. 22-11 ). The SCNIR analysis also identified a subgroup of patients with SCN at greatest risk for MDS/AML. The high-risk group with a G-CSF dosage of 8 µg/kg/day or more and ANC less than 2188 cells/µL had a cumulative incidence at 15 years of 34% for MDS/AML and 15% for death from sepsis, compared to 15% and 5%, respectively, for the low-risk group (G-CSF dosage <8 µg/kg/day and ANC ≥2188 cells/µL) (see Fig. 22-11 ). Importantly, however, even the patients with the best responses to G-CSF were not free of risk for MDS/AML or even death from sepsis. Although data were not available for blood counts at the time of onset of sepsis, these patients had demonstrated ANCs over 1000 cells/µL while receiving G-CSF therapy, suggesting that they were either not fully adherent to the treatment or that their granulocytes, although seemingly adequate in number, may have been limited in function. For patients in the “worst responder” category of high G-CSF dosage requirements and low ANC on therapy, early transplantation should be considered, depending upon donor availability, but the indication is not as strong as for those already showing abnormal cytogenetics.
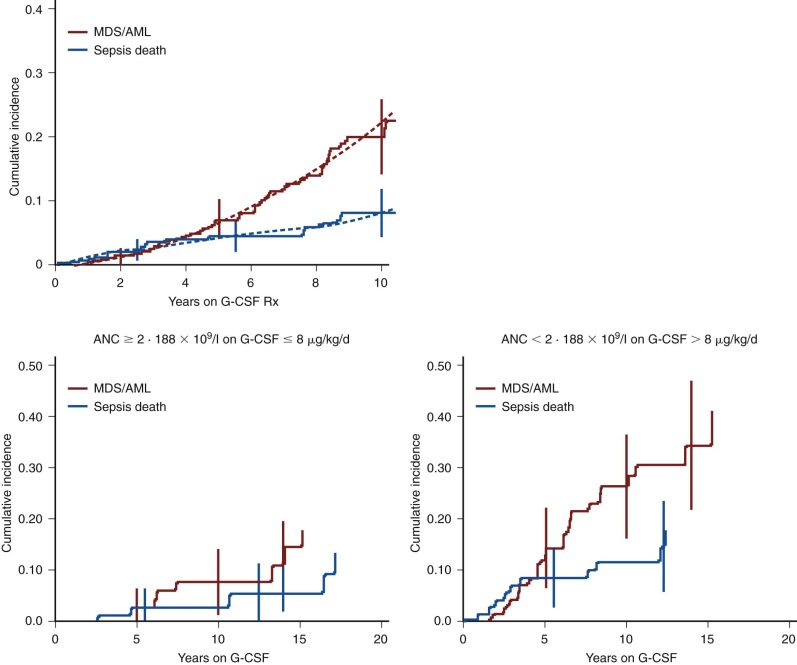
Analysis of ELANE gene mutations has also identified genetic risk factors for conversion to MDS/AML. In an SCNIR study of 188 patients with SCN and ELANE gene sequencing, 30 (16%) developed MDS/AML that was associated with 25 different mutations, but clusters of MDS/AML cases were associated with mutations C151Y, G214R, and S126L, and both these and other mutations identified in single MDS/AML cases tended to be associated with higher G-CSF dosage requirements and poorer ANC responses. On the other hand, there were no cases of MDS/AML in patients with mutations associated with both SCN and CyN phenotypes.
Cyclic Neutropenia.
CyN is an AD disorder characterized by regular periodic oscillations approximately every 21 days in the number of peripheral blood neutrophils, with a nadir of less than 200 cells per µL ( Fig. 22-12 ). The incidence of CyN has been estimated at 0.5 to 1 case per million population. During the neutropenic nadir of each cycle, patients may suffer from malaise, fever, oral and genital ulcers, gingivitis, periodontitis, and pharyngitis with local lymph node enlargement. Symptoms typically begin during the first year of life but may not present until adulthood. More serious complications can occasionally occur, including intestinal perforation with peritonitis, mastoiditis, and pneumonia. The severity of the infections tends to parallel the depth of the neutropenia, but some patients escape infections entirely during the neutropenic period. Many patients experience improvement in symptoms as they grow older. In these individuals, the cycles tend to become less noticeable as the neutrophil nadir rises to a more functionally adequate level. Although CyN is commonly viewed as a benign condition, 10% of patients in historical reviews have died of infectious complications. Pneumonias and peritonitis, the latter complicated by clostridial sepsis, have been the most common causes of death.
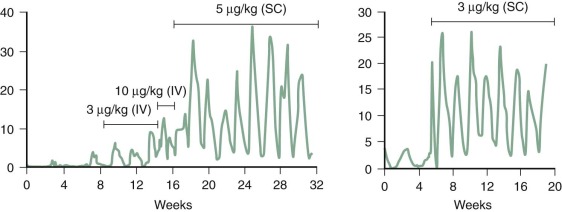
Oscillations in the rate of bone marrow production of neutrophils result in neutropenia with nadirs at intervals of 21 ± 3 days in the majority of patients. Neutrophil counts often fall to 0 at some time during the nadir and remain below 200 cells per µL for at least 3 to 5 days. In most patients, monocytosis occurs when neutrophil counts are at their lowest. In the recovery phase, the neutrophil counts rise, often into the low normal range, and monocyte counts drop. Oscillations are subtle in some individuals, particularly older patients, who instead have a pattern more consistent with chronic neutropenia. Oscillations in reticulocyte and platelet counts may parallel those seen for neutrophils, hence the alternative term cyclic hematopoiesis, but these other cell numbers range between normal and elevated levels and thus have no clinical impact. Marrow aspirates show highly variable morphology, depending upon the phase of the cycle. The myeloid series may show hypoplasia with “maturation arrest” at the myelocyte stage during the declining phase of the neutrophil oscillation, but normal or hyperplastic myelopoiesis during neutrophil recovery.
Patients with CyN have been shown to have mutations in the ELANE gene, most often in exons 4 or 5, but not other genes associated with SCN. How mutations in neutrophil elastase result in the cyclical production of neutrophils and other blood cells is not well understood. As discussed, both CyN and SCN are characterized by increased apoptosis of bone marrow neutrophil precursors, probably because of an excessive unfolded protein response to mutant elastase. Cycling may occur because of partial recovery of myelopoiesis stimulated by endogenous G-CSF production at the nadir, but with oscillation rather than homeostasis occurring as a result of the abnormal kinetics of the feedback loop.
CyN must be distinguished from cyclic fevers without neutropenia and from other forms of neutropenia, which can also produce less regular and lower amplitude variations in neutrophil counts. To establish the diagnosis of CyN, neutrophil counts should be monitored twice, or at least once, a week for 6 to 8 weeks. The genetic diagnosis can be confirmed by sequencing of the ELANE gene.
The management of patients with CyN includes careful attention to the identification and treatment of infections acquired when these patents are in a neutropenic phase. In general, fevers, upper respiratory symptoms, and cervical lymphadenopathy occurring during neutropenic episodes require no specific therapy. However, the physician must remain alert to symptoms suggesting a specific infection, particularly the sometimes fatal abdominal complications. Careful attention to oral and dental hygiene is important to minimize periodontal disease and ameliorate the discomfort of mouth sores. To prevent the development of severe infections and dental complications, G-CSF (filgrastim) therapy is recommended for patients with CyN with ANC nadirs consistently below 500 cells per µL.
G-CSF has been very effective in improving peripheral blood neutrophil counts in this disorder and alleviating the signs, symptoms, and risks associated with neutropenic nadirs. Most patients still experience cycles, and in fact the amplitude of the cycling may increase but with shortened cycle periods and increased neutrophil counts at the nadir (see Fig. 22-7 ). The dose of G-CSF required to maintain peripheral neutrophil counts in the normal range in most patients with CyN is usually 2 to 4 µg/kg/day, administered either daily or on alternate days. Cases of MDS or leukemia have not been reported for patients with CyN, including those treated long term with G-CSF.
Myelokathexis and WHIM Syndrome.
Myelokathexis is an uncommon form of moderate to severe chronic neutropenia characterized by granulocyte hyperplasia in the bone marrow, which contains degenerating neutrophils with cytoplasmic vacuoles, prominent granules, and nuclear hypersegmentation with very thin filaments connecting pyknotic-appearing nuclear lobes. Similar abnormalities can be seen in the majority of peripheral blood neutrophils. Eosinophils exhibit similar abnormal morphology, but lymphocytes, monocytes, and basophils appear normal, although relative lymphopenia has been noted in some patients. Recurrent warts and hypogammaglobulinemia (low IgG and occasionally low IgM and IgA) have often been reported in patients diagnosed with myelokathexis; hence the acronym WHIM.
Recurrent sinopulmonary infections and other bacterial infections in WHIM syndrome probably reflect an increased susceptibility because of both neutropenia and hypogammaglobulinemia. However, early deaths related to infection have not been reported, and affected patients are capable of mobilizing increased numbers of neutrophils into the peripheral blood during infectious episodes. Neutrophil function has generally been reported to be normal.
The genetic basis of WHIM syndrome was first identified as an AD truncating mutation in the cytoplasmic tail domain of the CXCR4 gene, encoding chemokine receptor-4, a G-protein–coupled receptor for CXCL12, (SDF-1). However, in other kindreds with wild-type CXCR4 genes and probable AR inheritance, including one with homozygous mutations in G6PC3 , patients’ leukocytes displayed similar marked enhancement of G-protein–dependent responses to CXCR4 ligands. Therefore the aberrant dysfunction of the CXCR4 -mediated signaling constitutes a common biologic trait of WHIM syndromes with different causative genetic anomalies. As the interaction between CXCR4 and CXCL12 provides a key retention signal for neutrophils in the bone marrow, myelokathexis has been suggested to result from retention of neutrophils in the marrow ( kathexis means retention ), where they undergo eventual apoptosis, evident in their diminished expression of the antiapoptotic factor bcl-x .
The neutropenia and excessive granulocyte apoptosis of WHIM syndrome are partially corrected by G-CSF or GM-CSF therapy, with responses typically seen within 4 to 8 hours of administration. Immunoglobulin levels also return to normal in patients treated with G-CSF. Specific therapy aimed at the primary molecular defect has recently been reported, using the CXCR4 antagonist plerixafor (formerly known as AMD3100 ) to correct the leukopenias of WHIM syndrome.
Shwachman-Diamond Syndrome.
Shwachman-Diamond syndrome, a rare multiorgan disease inherited as an AR trait, includes clinical findings of neutropenia, pancreatic exocrine insufficiency, short stature, and metaphyseal chondrodysplasia. This disorder is also described in Chapter 8 with a more detailed discussion of its genetics and pathophysiology. As in other bone marrow failure syndromes, patients are at risk for conversion to MDS/AML. The pancreatic acini are largely replaced by fatty tissue with relative sparing of the pancreatic ducts and islets. Growth failure and short stature are usually noted during the first or second year of life, and puberty is often delayed. Although the classic syndrome includes the triad of cytopenias, pancreatic dysfunction, and skeletal abnormalities, clinical presentations can be highly variable, including any number or none of these features.
Patients may show symptoms in infancy with eczema, skin infections, and otitis media; pneumonia, osteomyelitis, and sepsis can also be seen. Pancreatic insufficiency typically manifests in early infancy with steatorrhea, weight loss, and failure to thrive. Later in childhood, pancreatic function may improve, rendering the clinical diagnosis elusive. Levels of serum trypsinogen, pancreatic isoamylase, and stool elastase are generally low but not specifically diagnostic, and normal ranges are not well established for infants. Computed tomography scanning may reveal pancreatic lipomatosis. Mild abnormalities of the liver including hepatomegaly or elevated serum liver enzyme, are seen in 50% to 75% of younger patients, but they are usually mild and tend to resolve with age.
Common skeletal abnormalities include short stature and bone dysplasia evident in delayed appearance (but subsequent normal development) of secondary ossification centers as well as metaphyseal widening, first evident in the ribs (which appear short, with flared ends) and later in the proximal and distal femur. Other skeletal findings include clinodactyly, syndactyly, pes cavus, kyphosis, scoliosis, osteopenia, vertebral collapse, slipped femoral epiphysis, and supernumerary digits.
Neutropenia is the most common hematological manifestation of the syndrome, but some patients present with GI or skeletal problems and virtually normal blood counts. Neutrophil counts fall below 1000 cells per µL in approximately two thirds of patients, with no reciprocal monocytosis, but low counts may be intermittent. Some patients also have defects in chemotaxis or in numbers or function of B, T, and NK cells that may increase susceptibility to pyogenic infection. Shwachman-Diamond syndrome leukocytes may also show telomere shortening, which can overlap in severity with that observed in other bone marrow failure syndromes, even including dyskeratosis congenita. Anemia (normocytic or macrocytic) is common, and one fourth of patients have mild to moderate thrombocytopenia. Bone marrow studies have generally shown some degree of myeloid hypoplasia but are nondiagnostic.
The diagnosis of Shwachman-Diamond syndrome may be based on a combination of neutropenia, possibly other cytopenias, pancreatic insufficiency, and skeletal anomalies. Around 90% of patients who meet the clinical criteria harbor mutations in the Shwachman-Bodian-Diamond syndrome gene, SBDS , which functions in ribosomal biogenesis and other cellular processes. With increased detection of variant forms of the syndrome by genetic testing, a wider clinical phenotype has been recognized. In a study from the North American Shwachman-Diamond Registry, pancreatic lipomatosis was found in 22 of 23 patients, low fecal elastase in 14 of 17 patients, and low serum trypsinogen or pancreatic isoamylase in all 18 patients tested. Of 31 patients with clinical assessments, 26 had failure to thrive and 17 had diarrhea or steatorrhea; 17 had congenital anomalies. Neutropenia was the most common hematologic abnormality at presentation (77%), but two patients had isolated thrombocytopenia, three had severe anemia, and five had no cytopenias.
Treatment includes pancreatic enzyme replacement, which does not improve the neutropenia or dwarfism. Steatorrhea tends to diminish with time, although pancreatic insufficiency persists. The incidence of bacterial infections can vary among patients, generally depending upon the ANC. Administration of G-CSF increases the neutrophil count to the normal range and should be used in patients with persistently severe neutropenia or with recurrent or life-threatening infections.
Patients with Shwachman-Diamond syndrome may develop aplastic anemia, MDS, or leukemia, although usually later in life than patients with Fanconi anemia or SCN. These complications generally require treatment by HSCT. There are only very rare reports of solid tumors. A study from the French Severe Chronic Neutropenia Registry reported a 24% 20-year cumulative risk of aplastic anemia and MDS/AML in 102 patients; young age (<3 months) at first symptoms and low hematologic parameters both at diagnosis and during follow-up study were associated with severe hematologic complications. Because acquisition of a structural chromosomal abnormality, most commonly isochromosome i(7q) followed by del(20q), often precedes conversion to MDS, regular blood counts are recommended, and bone marrow examinations should be performed annually or more often in the face of evolving peripheral blood or cytogenetic findings. Cytogenetic abnormalities may be transient or unaccompanied by marrow dysplasia, so it remains unclear whether high-risk clonal aberrations constitute indications for HSCT.
Albinism-Neutropenia Syndromes
This group of rare primary immunodeficiency syndromes derive from AR defects in the biogenesis or trafficking of lysosomes and related late endosomal organelles. As a result, they share varying degrees of overlap in phenotype, including defects in formation of melanosomes (hence partial albinism), abnormal platelet function, and immunological defects involving not only neutrophil number but also the function of neutrophils, B lymphocytes, and cytotoxic T lymphocytes. They share a high risk of hemophagocytic lymphohistiocytosis (HLH) because of defects in T and NK cells. The partial albinism may be sufficiently subtle to require microscopic examination of hair for irregular, clumped melanosomes.
CHS is a rare genetic disorder characterized by partial oculocutaneous albinism, giant lysosomes in many cell types including granulocytes, and neuropathy. Most patients have moderate neutropenia, apparently because of ineffective granulopoiesis, and a high risk of HLH. CHS is described in detail later in this chapter.
Hermansky-Pudlak syndrome, another rare AR disease characterized by oculocutaneous albinism, is best known for associated platelet defects (see Chapter 8 ). One subtype, Hermansky-Pudlak syndrome type 2, also includes neutropenia and decreased numbers of NK cells, leading to increased susceptibility to infections and HLH. The AP3B1 gene responsible for this disorder, as well as for CyN in grey collie dogs, encodes an adapter protein involved in shuttling granule proteins, including neutrophil elastase, and thus may provide a connection to the molecular pathology of SCN and HLH.
Griscelli syndrome type 2, falls within the spectrum of Griscelli syndrome subtypes that share a phenotype of hypomelanosis with varying degrees of neurological impairment and immunodeficiency. The type 2 variant is characterized by neutropenia, hypogammaglobulinemia, partial albinism, and a predisposition to HLH. It is caused by mutations in the RAB27A gene, which encodes a small GTPase that serves as a key effector of granule exocytosis.
Cohen syndrome is an AR condition that includes neutropenia, pigmentary retinopathy, microcephaly, intellectual deficiency, and facial dysmorphism. The gene responsible for Cohen syndrome, COH1 (also termed VPS13B ), shares homology to a yeast protein that functions in vesicular sorting and intracellular protein trafficking. A case with congenital neutropenia, pigmentary retinopathy, and only subtle dysmorphism due to splicing mutations in VPS13B indicates a wider phenotype and a need to consider this gene in the evaluation of patients with neutropenia and retinopathy.
A single Mennonite family has been described with severe chronic neutropenia combined with short stature, albinism, coarse facial features, and recurrent bronchopulmonary infections by Streptococcus pneumoniae . This syndrome is caused by defects in the endosomal adaptor protein p14, which is involved in MAPK signaling to late endosomes.
Familial Benign Neutropenia
Familial benign neutropenia is a nonspecific descriptive diagnosis indicative of mild neutropenia with no tendency to increased infection. AD transmission has been identified in some familial cases. Although “familial benign neutropenia” has been described in Yemenite Jews, and in persons of German, French, American, and South African descent, these kindreds were investigated before the era of current immunological and molecular diagnostic tools, so they might now be classified with more specific diagnoses. For example, “benign ethnic neutropenia” in persons of African origin has recently been found to result from the Duffy negative genotype.
Other Causes of Neutropenia Caused by Intrinsic Defects in Myelopoiesis
Congenital or acquired bone marrow failure syndromes can occasionally present with isolated neutropenia. Fanconi anemia and dyskeratosis congenita should usually be recognized by other features associated with these disorders (see Chapter 8 ). Acquired aplastic anemia and MDSs may occur as primary processes, or as complications of bone marrow failure syndromes. Mild chronic neutropenia is a minor feature of monocytopenia and mycobacterial infection (MonoMAC) syndrome discussed later in this section.
Neutropenia Associated with Immune Dysfunction
Primary disorders of immunoglobulin production have been associated with neutropenic syndromes. One third of males with X-linked agammaglobulinemia have neutropenia at some time during the course of their disease. Persistent or CyN is common in patients with the hyper-IgM immunodeficiency syndrome, which in many instances appears to be secondary to the formation of autoantibodies. Immunoglobulin replacement therapy abolished the neutropenia in some but not all of these patients. AIN and other autoimmune cytopenias can also be seen in common variable immunodeficiency and isolated IgA deficiency.
Autoimmune lymphoproliferative syndrome (ALPS) reflects a family of defects in lymphocyte apoptosis. The syndrome includes lymphadenopathy, splenomegaly, and a variety of autoimmune disorders including immune thrombocytopenia, anemia, and neutropenia. Neutropenia associated with ALPS tends to be chronic, and because of the unique pathophysiology of the underlying disorder, represents one of the few forms of neutropenia treated by immunosuppression with corticosteroids or mycophenolate mofetil.
Cartilage hair hypoplasia, an AR disorder found in the Amish population, is characterized by short-limbed dwarfism, fine hair, moderate neutropenia (100 to 2000 cells per µL), and impaired cell-mediated immunity. The disorder arises from mutations in the noncoding RNA gene RMRP , which provides the RNA subunit of the pancreatic ribonuclease mitochondrial RNA processing enzyme, a complex involved in multiple cellular RNA processing events. G-CSF therapy has been reported to be effective in a single patient who also had an antineutrophil antibody ; bone marrow transplantation has corrected both the immunologic defects and neutropenia.
Schimke immuno-osseous dysplasia, a rare AR syndrome with variable expressivity, is characterized by spondyloepiphyseal dysplasia, steroid-resistant nephritic syndrome, lymphopenia with defective cellular immunity, neutropenia, and other cytopenias. Vasoocclusive processes, including cerebral and generalized atherosclerosis, are a life-limiting complication in the more severely affected patients. The disorder is associated with mutations in the gene SMARCAL1 , although the genetics may be heterogeneous. The immunodeficiencies and cytopenias can be treated with HSCT ; neutropenia, seen in 40% of patients, responds to G-CSF therapy.
Poikiloderma with neutropenia, Clericuzio type, is a rare AR genodermatosis characterized by early onset poikiloderma followed by pachyonychia, palmoplantar hyperkeratosis, and skeletal defects, as well as variable degrees of neutropenia and possible predisposition to malignancy. The gene mutated in this disorder, USB1 (also termed C16orf57 ), encodes a putative phosphodiesterase responsible for oligoadenylation of spliceosomal small nuclear RNA U6. Mutations in USB1 have also been identified in patients with Rothmund-Thomson syndrome (a phenotypically similar but distinct genodermatosis) and dyskeratosis congenita.
Neutropenia Associated with Metabolic Diseases
Significant neutropenia, often with relative monocytosis, occurs in Barth syndrome, a distinctive X-linked disorder caused by mutations in the TAZ gene, which encodes tafazzin, a phospholipid-lysophospholipid transacylase involved in the maintenance of mitochondrial cardiolipin. Clinical features include dilated cardiomyopathy, growth retardation, and 3-methylglutaconic aciduria. Affected boys develop symptoms in infancy or childhood, and neutropenia can precede the development of cardiac abnormalities. Neutrophil counts are variable and occasionally cyclic, ranging from nearly normal to very low, with recurring and sometimes life-threatening bacterial infections occurring in the most severely affected. Bone marrow shows maturation arrest at the myelocyte stage, with myeloblasts and promyelocytes demonstrating abnormal mitochondria.
Neutrophils in patients with glycogen storage disease type Ib (OMIM # 232220) are not only diminished in number, with neutrophil counts commonly less than 500 cells per µL, but also functionally defective, with abnormalities reported in chemotaxis, respiratory burst activity, and bacterial killing. The disorder derives from mutations in the glucose-6-phosphate transporter, encoded by the SLC37A4 gene. The defects in neutrophil survival and function are similar to those due to mutations in the myeloid-specific G6PC3 gene. Recombinant G-CSF has been effective in correcting the neutropenia and reducing serious infections in glycogen storage disease type Ib. Prolonged use of G-CSF has not been associated with any increased risk of MDS/AML in this disease.
Macrocytic anemia, often accompanied by neutropenia or thrombocytopenia, is a hematologic hallmark of Pearson syndrome, a rare and fatal congenital disorder involving not only the hematopoietic system but also the exocrine pancreas, liver, and kidneys. The bone marrow shows normal cellularity but striking abnormalities that include vacuolization of erythroid and myeloid precursors, hemosiderosis, and ringed sideroblasts. A mitochondrial defect related to large deletions in mitochondrial DNA, whose integrity depends on a specific DNA polymerase, likely leads to impaired hematopoiesis.
Neutropenia, usually accompanied by metabolic abnormalities including acidosis, ketosis, hyperammonemia, and hypoglycemia, can be a feature of organic acidemias such as propionic acidemia, methylmalonic acidemia, methylmalonic aciduria, and others. Although these congenital disorders are usually evident in the first 10 days of life, variants can present in older children or adolescents. Diagnosis is based on screening of urine organic acids, followed by more specific metabolic or genetic testing. Disorders of cobalamin metabolism can present in the first months to years of life with neutropenia and are usually accompanied by macrocytic anemia and often by methylmalonic acidemia.
Neutropenia Caused by Extrinsic Factors
Infection.
The most common cause of transient neutropenia in childhood is viral infection. Viruses commonly causing acute transient neutropenia include influenza A and B, adenovirus, respiratory syncytial virus, enteroviruses, human herpes virus 6, measles, rubella, and varicella. Neutropenia develops during the first 24 to 48 hours of the illness and may persist for 3 to 6 days. This period usually corresponds to the time of acute viremia and may relate to virus-induced redistribution of neutrophils from the circulating to the marginated granulocyte pool, sequestration, or increased neutrophil utilization after tissue damage caused by the viruses.
Both transient and chronic neutropenia occur with Epstein-Barr virus infection and may be related to splenomegaly, direct infection of progenitors, and antineutrophil antibodies. Cytomegalovirus (CMV) infection and anti-CMV therapies may cause neutropenia in the setting of perinatal infection or immune suppression (particularly HSCT or solid organ transplant) but can also affect the immunocompetent. Parvovirus B19 and hepatitis A and B may also cause neutropenia, although they are more commonly associated with pure red cell aplasia and multiple cytopenias, respectively.
Leukopenia, either transient or chronic, is also common in patients with HIV infection as a result of multiple causes, including antiretroviral drugs, cellular immune dysfunction, ineffective hematopoiesis, antineutrophil antibodies, B 12 or folate deficiency, and hypersplenism.
Significant neutropenia may occur during typhoid, paratyphoid, tuberculosis, brucellosis, tularemia, and rickettsial infections. The mechanisms responsible for neutropenia in these conditions remain ill-defined. During periods of relapsing fever caused by acute malaria, the apparent neutropenia may be secondary to increased neutrophil margination in the spleen.
Neutropenia in patients with bacteremia and endotoxemia may result from excessive destruction of neutrophils with depletion of the bone marrow reserve pool and constitutes a dire prognostic sign, particularly in newborns. This consumption can occur after phagocytosis of microbes, from the release of metabolites of arachidonic acid, or from activation of the complement system through either the alternate or classic pathway. The resultant generation of the chemoattractant C5a induces neutrophil aggregation and leads to the formation of leukoemboli, which adhere to and damage endothelial surfaces in the pulmonary capillary bed. Complement-mediated neutropenia may also occur transiently during hemodialysis, during continuous-flow leukapheresis, and after burn injury.
Newborns with sepsis may benefit from granulocyte transfusion or G-CSF, but clinical trials have not yet provided compelling evidence for benefit from either modality.
Drug-Induced Neutropenia.
Drug-induced neutropenia, defined as an idiosyncratic reaction to the offending drug, excludes disorders in which other cell lines are perturbed (e.g. drug-related aplastic anemia) and the predictable neutropenias observed with anticancer therapy. Idiosyncratic reactions tend to develop more often in women than in men and more often in older than in younger persons.
In its most serious form, drug-induced agranulocytosis, neutrophil counts reach levels below 200 cells/µL, with high morbidity (e.g. deep tissue infection and sepsis) and mortality rates as high as 5%. Most deaths occur in patients who are elderly or have metabolic complications such as shock or renal failure.
Almost any drug can cause neutropenia; Table 22-9 lists the agents most commonly associated with isolated, idiosyncratic neutropenia or agranulocytosis. Drugs reported to have been associated with agranulocytosis have been extensively reviewed. The most common drug classes include antiinflammatories, particularly aminopyrine and sulfasalazine; antimicrobial agents, particularly trimethoprim and sulfamethoxazole; antithyroid drugs; antipsychotics such as clozapine and phenothiazines; and others such as deferiprone and rituximab (see Table 22-9 ).
| Possible Mechanism | |||
|---|---|---|---|
| Drug | Direct Suppression | Metabolite Suppression | Immune Destruction |
| Antiinflammatory | |||
| Aminopyrine | X | ||
| Ibuprofen | X | ||
| Indomethacin | X | ||
| IV immunoglobulin | X | ||
| Phenylbutazone | X | ||
| Sulfasalazine | X | ||
| Anticonvulsant | |||
| Carbamazepine | X | ||
| Phenytoin | X | ||
| Valproic acid | X | ||
| Antimicrobial | |||
| Chloramphenicol | X | ||
| Dapsone | X | ||
| Penicillins | X | X | |
| Sulfonamides | X | ||
| Trimethoprim/Sulfa | X | ||
| Antithyroid | |||
| Methimazole | X | ||
| Propylthiouracil | X | ||
| Cardiovascular | |||
| Hydralazine | X | ||
| Procainamide | X | ||
| Quinidine | X | ||
| Psychotropic | |||
| Chlorpromazine | X | ||
| Clozapine | X | ||
| Olanzapine | X | X | |
| Phenothiazines | X | ||
| Other | |||
| Chlorpropamide | X | ||
| Cimetidine, ranitidine | X | ||
| Deferiprone | |||
| Gold | X | ||
| Levamisole | X | ||
| Rituximab | |||
| Ticlopidine | X | ||
* Partial list of agents capable of causing idiosyncratic drug-induced neutropenia. Italics indicate the drugs with the highest relative risk of neutropenia and agranulocytosis. The mechanism, when unknown, has been left blank; some of the indicated mechanisms are not well established.
Although the underlying mechanisms for most drug-induced neutropenias are unknown, most studies suggest that they can be classified as leading to toxic suppression of neutrophil formation or immune destruction of mature cells. Neutropenia resulting from myelosuppression is usually insidious in onset and may be asymptomatic. Oral mucositis may be the first clinical sign before infectious complications. Individual differences in drug pharmacokinetics can lead to toxic levels of the drug or metabolites in the marrow microenvironment. For example, whereas neutropenia can be observed in any patient taking sulfasalazine, individuals who are slow acetylators show much greater toxicity than do those who are fast acetylators. Alternatively, a susceptible patient’s myeloid precursors may be abnormally sensitive to typical drug concentrations, as observed in neutropenia that can appear 20 to 40 days after starting phenothiazines.
Drugs can also induce immune-mediated peripheral destruction of neutrophils by several mechanisms. In one, the drug serves as a hapten in promoting the synthesis of antibodies that are capable of destroying mature neutrophils. Aminopyrine, penicillin, propylthiouracil, and gold can cause neutropenia by this mechanism. Alternatively, the drug can elicit the formation of circulating immune complexes that attach to the surface of the neutrophil and lead to its destruction, as in the case of quinidine. In addition, immunologic changes induced by drugs can suppress granulopoiesis. For example, activation of both cellular and humoral immune responses has been reported to impair myelopoiesis after therapy with quinidine and phenytoin, respectively. Immune-mediated neutropenia is characterized by abrupt onset, often with fever, chills, and prostration, occurring 7 to 14 days after the first use of the drug or immediately after reexposure. Transient, usually mild, neutropenia may follow treatment of immune thrombocytopenia with intravenous immunoglobulin.
The duration of drug-induced neutropenia is highly variable. Myelosuppressive drug reactions may last from a few days to months or years, whereas immune-mediated reactions usually last for 6 to 8 days. Once neutropenia occurs, the most important therapeutic action is, of course, to withdraw all drugs that are not absolutely essential, particularly those most highly associated with neutropenia. G-CSF therapy may hasten neutrophil recovery slightly, but evidence-based data are lacking to justify its routine use ; therefore the clinical indication is questionable unless the patient has a severe infection. During recovery from drug-induced neutropenia, with or without G-CSF therapy, rebound leukocytosis can occur and is accompanied by a left shift in the peripheral blood and bone marrow myeloid series with occasional circulating immature leukocytes.
Autoimmune Neutropenia.
AIN can occur as an isolated process, as a manifestation other autoimmune diseases, or as a secondary complication of infection, drugs, or malignancy. In primary AIN, low circulating neutrophil counts are the only hematological finding, and associated diseases or other factors that cause neutropenia are absent. The peak incidence of AIN occurs in infants and young children, where it is a generally benign disorder that remits spontaneously, as discussed in the following section. Many cases of chronic idiopathic neutropenia in children and adults are now recognized as secondary to more generalized autoimmune disorders ( Box 22-5 ).
AUTOIMMUNE DISORDERS
Autoimmune cytopenias (Evans syndrome)
Autoimmune lymphoproliferative syndrome
Felty syndrome (arthritis, splenomegaly, leukopenia)
Primary biliary cirrhosis
Sjögren syndrome
Scleroderma
Systemic lupus erythematosus
INFECTION
Infectious mononucleosis
Human immunodeficiency virus
MALIGNANCY
Leukemia
Lymphoma (Hodgkin and non-Hodgkin)
HYPOGAMMAGLOBULINEMIAS, DYSGAMMAGLOBULINEMIAS
Common variable immunodeficiency
Hyperimmunoglobulin M syndrome
Immunoglobulin A deficiency
X-linked agammaglobulinemia
ANGIOIMMUNOBLASTIC LYMPHADENOPATHY (CASTLEMAN DISEASE)
DRUG REACTION (see Table 22-9 )
Patients with AIN may have highly (and rapidly) varying neutrophil counts, ranging from the mild neutropenia to agranulocytosis. Monocytosis is common. Bone marrow examination generally shows normal to increased cellularity with myeloid hyperplasia and normal to increased numbers of mature neutrophils, although diminished neutrophils or maturation arrest at earlier stages of differentiation can also be seen. Mild splenomegaly is occasionally present. The incidence of pyogenic infection is not always related to the degree of neutropenia and is generally limited to cutaneous and respiratory infections. Serious infections are generally uncommon. Spontaneous remissions resolve the process in most infants, but occur less commonly in older children (discussed later).
Neutrophil-specific cell surface antigens identified as targets of autoantibodies include human neutrophil antigen (HNA) 1a, formerly termed NA1 ; HNA-1b, formerly NA2; and HNA-2, formerly NB1. The HNA-1a and HNA-1b antigens are glycosylated isoforms of the neutrophil immunoglobulin receptor FcγIIIb (CD16) (see Table 22-6 ), and the HNA-2 antigen is CD177, the counter-receptor for PECAM-1. Mechanisms that trigger autoantibody production are unknown. In children, primary AIN is usually associated with HNA-allele–specific antibodies, but secondary AIN may be associated with pan-FcγRIIIb antibodies.
The neutropenia of AIN is presumed primarily to be caused by peripheral destruction of antibody-coated neutrophils, which may be also be augmented by the deposition of C3. Phagocytosis of neutrophils in the spleens of patients with AIN has been observed. In some cases, antineutrophil antibodies also appear to interfere with myelopoiesis. Impairment of phagocytosis, respiratory burst activity, and adhesion by neutrophil-directed antibodies has also been observed, which may contribute to the risk of infection in AIN.
Antineutrophil antibodies can be detected by direct or indirect granulocyte immunofluorescence tests (analogous to the direct and indirect antiglobulin tests for erythrocyte antibodies but usually performed by flow cytometry) or by granulocyte agglutination tests. Granulocyte agglutination is less sensitive but may be useful for detecting neutrophil-binding immune complexes in Felty syndrome. All of these tests are subject to false positive results from anti-HLA antibodies or nonspecific binding of immunoglobulins to neutrophil FcRs, as well as false negative results caused by high background levels of bound immunoglobulins. Because of the limited sensitivity and specificity of these assays, even in combination, laboratory tests for antineutrophil antibodies are neither necessary nor sufficient to make the diagnosis of AIN, and their interpretation depends on the specific clinical setting.
Importantly, the standard laboratory test for antineutrophil cytoplasmic antibody does not detect antibodies responsible for AIN.
Treatment of patients with immune neutropenia includes the judicious use of appropriate antibiotics for bacterial infections, plus antimicrobial mouthwashes and regular dental hygiene for mouth sores or gingivitis. Infections tend to be less common in immune neutropenia than with the corresponding degree of neutropenia from other causes, probably because granulopoiesis is in most cases intact and tissue delivery of neutrophils is greater than that with equivalent neutrophil levels caused by impaired production. Prophylactic administration of trimethoprim/sulfamethoxazole may be also helpful for management of recurrent minor infections, although there are no controlled studies addressing the efficacy of this approach.
For definitive therapy, administration of G-CSF starting at low dosages of 1 to 2 µg/kg/day has been successful in increasing the neutrophil count to the normal range or even higher in patients with primary AIN, including secondary immune neutropenia and Felty syndrome. The initial response is usually apparent within days of starting therapy but may require up to 2 weeks. Patients with normal or high ANC during therapy may be weaned to a very low dosage or administration of G-CSF on alternate days. The incidence of bone pain appears to be higher in AIN than in hypoproductive neutropenia, probably because of expansion of the already hyperplastic bone marrow. Because most patients with immune neutropenias do not suffer serious infectious complications, G-CSF therapy should be reserved for the few with serious or recurrent infections or with multiple hospitalizations for febrile neutropenia. When used for AIN and other forms of acquired neutropenia, G-CSF is not associated with development of MDS/AML.
Intravenous immunoglobulin administration can result in normalization of the neutrophil count, but efficacy is variable and often short-lived. Corticosteroids can raise the neutrophil count but may actually increase the risk of serious infection. Similarly, splenectomy can provide transient benefit that is offset by the risk of sepsis, and the procedure is limited to conditions with clinically significant splenomegaly such as Felty syndrome.
T-cell large granular lymphocyte leukemia (LGL), a clonal disorder of cytotoxic T lymphocytes most common in the elderly, often presents as severe chronic neutropenia with or without accompanying anemia. LGL-associated neutropenia, which overlaps clinically with Felty syndrome, may derive from either immune complex– or cell-mediated mechanisms and generally responds to a combination of immunosuppressive and cytotoxic therapy for the underlying disease and G-CSF for the neutropenia.
Neutropenia can be seen as part of a broader spectrum of disease in a variety of other acquired disorders of the immune system. Autoimmune diseases such as systemic lupus and rheumatoid arthritis can be associated with antibody or immune complex-mediated destruction of neutrophils or their precursors. Felty syndrome refers specifically to neutropenia in association with splenomegaly and seropositive rheumatoid arthritis; there is considerable pathophysiologic overlap between Felty syndrome and rheumatoid arthritis–associated LGL. Therapy with G-CSF is effective but may precipitate flares of the underlying disease ; splenectomy may be effective, as discussed previously.
Chronic Benign Neutropenia and Autoimmune Neutropenia of Childhood.
Most cases of what was termed chronic benign neutropenia of infancy and childhood are now believed to represent an AIN that has parallels to childhood idiopathic thrombocytopenic purpura. AIN of childhood occurs predominantly in children younger than 3 years ( Box 22-6 ). The median age of presentation is 8 to 11 months, with a range of 2 to 54 months. There is a slight female predominance. Although AIN of childhood is the most common cause of chronic neutropenia in the pediatric age group, it is still a relatively uncommon diagnosis, with an incidence of approximately 1 in 100,000 children per year. However, this figure is probably an underestimate because of the generally benign course of this disorder.
Considerable Overlap with Chronic Benign Neutropenia of Childhood
| Incidence | Most common cause of chronic neutropenia in infancy and childhood; incidence ≥1 per 100,000 children per year. |
| Clinical features | Median age of diagnosis is 8-11 months of age (range 3-38 months). |
| Relatively minor infections (otitis media, gingivitis, upper respiratory tract, skin) occur with an increased frequency only in some patients and respond well to antimicrobial therapy. Rare patients (usually young infants) have pneumonia or sepsis. | |
| Laboratory evaluation | Median absolute neutrophil count at time of diagnosis ≈200 neutrophils/µL (range 0-500 cells/µL). Hemoglobin, platelet count generally normal. Antineutrophil antibodies can be detected in many patients and are often directed to human neutrophil antigen (HNA) 1 (FcγRIII). Bone marrow test (if performed) shows normal to increased myelopoiesis, sometimes with a decrease in mature neutrophils or maturation arrest at earlier stages. |
| Therapy | Antibiotics for acute infection; prophylactic antibiotics may be helpful in some patients with recurrent otitis media. Consider use of granulocyte colony-stimulating factor only in the event of serious infection, very frequent minor infections, or very frequent precautionary emergency room visits or admissions. |
| Prognosis | Excellent. Although the ANC often remains below 500 cells per µL for 12 or more months, spontaneous remission occurs in almost all patients (median of 20 months, range 6-54 months). |
The presenting ANC is often in the severe range (<200 cells/µL), usually with a relative monocytosis and/or eosinophilia. Transient increases in circulating neutrophil counts can occur in response to infection. Other peripheral counts are normal for age. Mild thrombocytopenia has been reported, but the presence of other autoimmune cytopenias more likely represents Evans syndrome (see Chapter 13 ), which is more likely to become chronic and to be associated with underlying immunodeficiency disorders or ALPS.
Antineutrophil antibodies can be detected in many patients by repeated testing with a combination of immunofluorescent and agglutination assays, but absence of detectable antibody does not exclude the diagnosis, and a positive result does not exclude congenital neutropenia. Bone marrow examination generally shows normal to increased cellularity with myeloid hyperplasia and normal to increased numbers of mature neutrophils.
Bacterial infections in AIN of childhood are generally mild (e.g., skin infections, otitis media, gingivitis) and responsive to standard antibiotics. Cellulitis involving the labia majora was seen in 23% of 26 girls with AIN in one series, often the result of Pseudomonas aeruginosa . Pneumonia, sepsis, and meningitis have been reported only occasionally in infants with AIN. Spontaneous remission occurs in almost all patients, with neutropenia persisting for a median duration of 20 months. Children younger than 9 months of age at the time of diagnosis may recover normal peripheral neutrophil counts more rapidly. With increasing age, spontaneous remission becomes less likely.
Febrile episodes should be managed aggressively in the initial period before the diagnosis of childhood AIN is established, including the use of broad-spectrum parenteral antibiotics. However, once the diagnosis is definite and the patient has had no serious infections, subsequent febrile illnesses can be managed more conservatively if clinically warranted. Prophylactic trimethoprim/sulfamethoxazole can be helpful for children with recurrent otitis media and other minor infections. Administration of G-CSF results in an increased peripheral neutrophil count in childhood AIN and is the treatment of choice for the rare child with AIN who manifests very severe or recurrent major infections. G-CSF doses of 1 to 2 µg/kg/day or less are usually effective, and responses are typically seen within days. Steroids and intravenous gamma globulin have also been used, but have limited efficacy, as discussed above.
Neonatal Immune Neutropenias
Immune-mediated neutropenia in the newborn may be isoimmune because of maternal immune response to fetal alloantigens or maternal autoimmune as a result of passive transfer of maternal autoantibody.
Neonatal isoimmune neutropenia, an immunologic disorder analogous to Rh hemolytic disease, results from maternal sensitization to fetal neutrophils bearing antigens that differ from the mother’s. Maternal IgG antibodies cross the placenta and result in an immune-mediated neutropenia that can be severe and lasts from several weeks to as long as 6 months. The infant bone marrow demonstrates myeloid hyperplasia, usually with depletion of mature neutrophil forms.
Neutrophil antibodies are found in the serum of the mother and the infant and are often directed to the neutrophil-specific HNA antigen system. The HNA-1 antigens most often responsible are isotypes of the neutrophil Fcγ receptor IIIb (see Table 22-6 ). Affected infants can be asymptomatic or develop omphalitis, cutaneous infections, pneumonia, sepsis, and meningitis. Since neonatal sepsis can be itself associated with profound neutropenia, the underlying immune-mediated neutrophil destruction may not be appreciated immediately in affected newborns who have signs of sepsis.
The initial management of neonatal isoimmune neutropenia should include parenteral antibiotics, even if signs of infection are absent, because of the association of neutropenia with neonatal sepsis. Intravenous γ globulin may sometimes be effective in increasing the neutrophil counts. Treatment with G-CSF starting at 5 to 10 µg/kg/day intravenously should be considered for very profound neutropenia or when serious infections develop in infants with isoimmune neutropenia.
Neonatal immune neutropenia can also occur in infants of mothers with AIN. Transplacental passage of an IgG antineutrophil antibody can react with the infant’s neutrophils bearing the inciting antigen and result in a profound neutropenia lasting 2 to 4 weeks. Treatment is similar to that for isoimmune neutropenia. Leukoagglutinating antibodies directed against HLA antigens are commonly found in multiparous women and their newborn children’s sera ; however, these antibodies are not associated with neutropenia and thus need to be distinguished from neutrophil-specific antibodies in laboratory assays.
Nutritional Deficiencies.
Ineffective granulopoiesis is part of the megaloblastic marrow pathology observed in patients with nutritional deficiencies of vitamin B 12 or folic acid. Neutropenia also occurs with severe protein-calorie malnutrition, as in anorexia nervosa and marasmus. Neutropenia and marrow megaloblastosis have also been observed in copper deficiency Notably, these nutritional deficiencies do not cause isolated neutropenia; anemia is virtually universal, and thrombocytopenia is an occasional additional finding.
Reticuloendothelial Sequestration.
Splenic enlargement caused by portal hypertension, intrinsic splenic disease, or splenic hyperplasia can lead to neutropenia that is usually accompanied by similar degrees of thrombocytopenia and anemia. The neutropenia is usually mild and may be ameliorated by successful treatment of the underlying disease. In selected situations, splenectomy may be necessary to restore the neutrophil count to normal.
Bone Marrow Infiltration.
Malignancies such as leukemia, lymphoma, and solid tumors that infiltrate the bone marrow result in a myelophthisic picture, producing leukoerythroblastic peripheral blood smears with neutropenia that is usually accompanied by other cytopenias. Tumor-induced myelofibrosis may further reduce the peripheral blood neutrophil count. Neutropenia can also result from bone marrow involvement in granulomatous infections, lysosomal storage disease, or osteopetrosis.
Chronic Idiopathic Neutropenia
Chronic idiopathic neutropenia represents a group of disorders that by definition are poorly understood and cannot be placed in any of the previously named categories. Investigation of the mechanism of neutropenia has suggested that in some cases, decreased or ineffective production of neutrophils may be the result of excessive apoptosis, such as in congenital neutropenia. Overall, the clinical features, bone marrow findings, and natural history of chronic idiopathic neutropenia are variable but generally milder than congenital neutropenia; it is likely that many of the cases described in older studies had antibody-mediated neutrophil destruction and normal bone marrow reserve. Treatment, when necessary, should be based on the severity of symptoms; G-CSF has proven effective at lower doses than in SCN and without any reported increase in risk of MDS/AML.
Evaluation of the Patient with Neutropenia
The basic approach to a patient with neutropenia ( Fig. 22-13 ) includes a history and physical examination with emphasis on: (1) related phenotypic abnormalities; (2) determination of whether bacterial infection is present (including evaluation of gingiva and perineum); (3) evaluation of lymphadenopathy, hepatosplenomegaly, and any other signs of an underlying associated chronic illness; and (4) history of recent infection and drug exposure. The frequency and duration of symptoms is important, and a history of periodontitis, dental abscesses, or tooth loss is particularly suspicious for significant chronic or recurrent neutropenia. The family history may reveal other individuals with recurrent infection. Unexplained deaths and the ethnic background in children younger than 1 year of age should be noted.
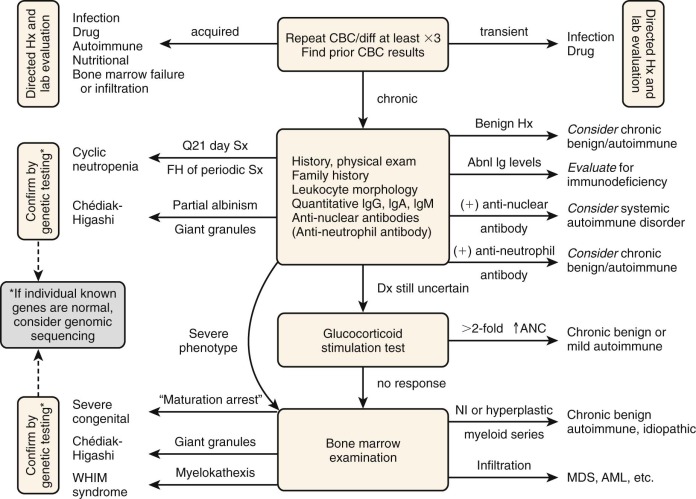
The duration and severity of the neutropenia and the presence or absence of significant other symptoms or physical findings greatly influence the speed and extent of laboratory evaluation. If the patient has isolated neutropenia, is asymptomatic, and other findings are absent, clinical observation for several weeks is usually the best approach. Any medications known or suspected to be associated with neutropenia should be discontinued. Any signs of an acute bacterial infection in a patient with moderate to severe neutropenia and fever calls for prompt evaluation and treatment, including the use of intravenous broad spectrum antibiotics.
Patients with persistent neutropenia should have white blood cell and differential counts obtained at least twice weekly for 6 to 8 weeks in order to evaluate for the periodicity of CyN. Direct and indirect antiglobulin tests should be performed to evaluate for the presence of red cell autoantibodies and quantitative serum immunoglobulins (IgG, IgA, and IgM, but not IgE) measured to detect associated immunoglobulin abnormalities. HIV testing is also indicated in chronic cases. Antineutrophil antibody testing, although technically imperfect (as discussed), is generally included in an initial evaluation because of the frequency of AIN.
Bone marrow aspiration and biopsy with cytogenetics should be part of the initial evaluation if leukoerythroblastosis, anemia, macrocytosis, or thrombocytopenia is present and should be performed eventually if the diagnosis remains obscure despite initial testing. If SCN is suspected, bone marrow analysis with cytogenetic screening for monosomy 7 should be performed both to establish the diagnosis (through demonstration of “maturation arrest”) and to rule out MDS before G-CSF therapy. Other laboratory tests that should be considered, depending on the clinical situation, include evaluation for collagen vascular disease, exocrine pancreatic insufficiency, metabolic disorders (e.g. organic acidurias), and measurement of B 12 , folate, and copper levels. Radiographic studies may be useful in the diagnosis of Shwachman-Diamond syndrome. Assessing the ANC before and 4 to 6 hours after a single dose of glucocorticosteroid (usually prednisone 1 to 2 mg/kg) measures mobilization of the bone marrow reserve pool of mature neutrophils ; an increase in the ANC to a normal or only moderately low level indicates “chronic benign” or idiopathic neutropenia and may render bone marrow examination unnecessary. The identification of genes responsible for most forms of congenital neutropenias (discussed previously) now permits specific genetic diagnosis in many cases and provides a basis for genetic counseling and prenatal diagnosis.
Principles of Therapy for Neutropenia
The management of neutropenia depends on the underlying cause and severity of the neutropenia. The major concern in patients with neutropenia is the development of serious pyogenic infection. Patients with severe neutropenia (ANC ≤500 cells/µL) with poor marrow reserves are at highest risk for developing progressive infection and septicemia. Fever may often be the only indication of infection, because local signs and symptoms of inflammation may be diminished in the face of neutropenia. Organisms involved are usually from the skin or GI tract. Thus febrile patients with severe neutropenia caused by poor marrow function should be treated promptly with broad-spectrum antimicrobials after blood and other appropriate cultures are obtained, and treatment should be continued as long as the patient is febrile, even if blood cultures are negative. Patients with neutropenia who are febrile for more than 4 days while receiving broad spectrum antibacterial antibiotics are at risk for fungal infection, so empiric treatment with antifungal antibiotics should be considered in this setting. Patients who have neutropenia with documented fungal infection or gram-negative bacterial sepsis who have not responded to appropriate therapy are candidates for granulocyte transfusions.
Patients with neutropenia and normal to increased marrow cellularity (e.g., in the setting of AIN) often have a minimally increased risk of pyogenic infection and respond more briskly to appropriate antibiotics. A less aggressive course may be a reasonable approach to a febrile illness in such patients who appear clinically well, even if the ANC is below 500 cells/µL. A child with AIN who develops fever or infection can generally be managed as an outpatient unless the infection is severe.
Chronic gingivitis and periodontitis can be a persistent source of morbidity and result in tooth loss in patients with chronic or CyN. Good dental hygiene and regular professional treatment are necessary to forestall or prevent tooth loss and far outweigh any theoretical risk of bacteremia from local trauma.
The advent of recombinant G-CSF and other hematopoietic growth factors has revolutionized the therapy of neutropenias. As discussed in previous sections, G-CSF has been successfully used to increase the neutrophil count in a wide variety of conditions including SCN, CyN, and immune-mediated neutropenias. Continuous G-CSF is recommended for all patients with SCN and other conditions with poor marrow production of neutrophils and a high risk of developing serious infection. For disorders with lower risks of infection, such as autoimmune or drug-induced neutropenia, G-CSF therapy should be reserved for specific clinical indications such as serious or repeated infections. In general, more frequent administration (daily or alternate days) of the lowest dose needed to achieve an ANC greater than 1000 cells/µL produces a more steady response and lower toxicity than less frequent administration (e.g., weekly) of higher doses. Polyethylene glycol-filgrastim has been used off label in a few patients who were unresponsive to filgrastim, but it causes considerable bone pain and more serious toxicities.
Antibiotic prophylaxis, usually with trimethoprim-sulfa, may be useful for some patients with neutropenia and normal to increased myelopoiesis who do not meet criteria for G-CSF therapy. There are no evidence-based data to indicate the effectiveness of measures beyond careful handwashing to prevent infections, most of which arise from the patients’ own skin and gut flora; there is no need for reverse precautions or neutropenic diets in patients who have neutropenia without concurrent immunosuppression. To avoid invasive fungal infection, patients with chronic neutropenia should also avoid mulch, dusty construction or demolition sites, and bird or animal waste.
Neutrophilia
Neutrophilia refers to an alteration in the total number of blood neutrophils that is in excess of about 7500 cells/µL in children and adults. During the first few days of life the upper limit of the normal neutrophil count ranges from 7000 to 13,000 cells/µL for neonates born prematurely and at term gestation, respectively, and is followed by a decrease to adult levels within the first few weeks of life. An increase in circulating neutrophils is the result of a disturbance of the normal equilibrium involving neutrophil bone marrow production and movement in and out of the marrow and circulation. Three primary mechanisms accounting for neutrophilia act most often in combination ( Box 22-7 ). First, increased bone marrow production can expand the circulating neutrophil pool through: (1) increased progenitor cell proliferation and terminal differentiation through the neutrophilic series; (2) increased mitotic activity of neutrophilic cell precursors; or (3) shortening of the cell cycle time in the mitotic pool of neutrophil precursors. Second, increased numbers of neutrophils may be mobilized from either the bone marrow storage compartment or the peripheral marginating pools into the circulating pool. Additionally there may reduced neutrophil margination or egress into tissue.
Increased Production
Chronic infection
Chronic inflammation
Ulcerative colitis
Rheumatoid arthritis
Tumors
Postneutropenia rebound
Myeloproliferative disease
Drugs (lithium; rarely ranitidine, quinidine)
Chronic idiopathic neutrophilia
Familial cold urticaria
Enhanced Release From Marrow Storage Pool
Corticosteroids
Stress
Hypoxia
Acute infection
Endotoxin
Reduced Margination
Stress
Infection
Exercise
Epinephrine
Leukocyte adhesion deficiency
Decreased Egress From Circulation
Corticosteroids
Splenectomy
Leukocyte adhesion deficiency
Acute neutrophilia occurs rapidly within minutes in response to exercise or epinephrine-induced reactions and has been attributed to mobilization of the marginating pool of neutrophils into the circulating pool. Slower onset of acute neutrophilia can occur after glucocorticoid administration or with inflammation or infection associated with the generation of endotoxin, TNF, IL-1, IL-8, and hematopoietic growth factors such as G-CSF. Maximal response usually occurs within 4 to 24 hours after exposure to these agents and is probably the result of release of neutrophils from the marrow storage compartment into the circulation. The mechanisms that underlie release from the marrow pool involve interactions between neutrophils and the bone marrow stroma mediated by adhesion molecules (such as β2 integrins) and by chemokines such as CXCL12 and its receptor CXCR4. Glucocorticoids may also slow the egress of neutrophils from the circulation into tissue.
Chronic neutrophilia may accompany the prolonged administration of glucocorticoids, persistent inflammatory reactions, infection, chronic blood loss, or chronic anxiety. Marked neutrophilia is a hallmark of functional disorders of neutrophils that are caused by impaired adhesion or motility, such as in patients with leukocyte adhesion deficiencies or actin dysfunction (see later). Sustained moderate neutrophilia invariably follows either surgical or functional asplenia, probably because of decreased clearance of circulating neutrophils. Neutrophilia also occurs in familial cold urticaria and periodic fever syndromes.
Congenital primary neutrophilia is rare. In one family with AD hereditary neutrophilia, affected individuals maintained ANCs of 14,000 to 164,000 cells/µL along with hepatosplenomegaly, increased leukocyte alkaline phosphatase, and Gaucher-type bone marrow histiocytes. A more recently-described AD neutrophilia kindred of 12 patients in three generations had an activating mutation in the CSF3R gene leading to constitutive activation of the G-CSF receptor and increased proliferation and differentiation of neutrophil precursors. Affected family members had a median neutrophil count of 16,900 cells/µL (range: 11,000 to 23,700) and one progressed to MDS.
Leukemoid Reactions
Leukocyte counts greater than 50 × 10 3 normal cells/µL are referred to as leukemoid reactions. The peripheral blood may show small proportions of immature myeloid cells, including occasional myeloblasts and promyelocytes. Leukemoid reactions need to be distinguished from chronic myelocytic leukemia (CML), which features a more extreme “left shift” and basophilia in the peripheral blood differential count, as well as the characteristic clinical, laboratory, and cytogenetics signs of a myeloproliferative disorder (see Chapter 11 ). Chronic neutrophilic leukemia is a distinct, extremely rare, malignant cause of mature neutrophilia, with a mean age at diagnosis of 62.5 years (range 15 to 86) in 33 reported cases.
Leukemoid reactions are most commonly triggered by pyogenic infections, especially by S. aureus or S. pneumoniae but can also occur with other infections such as tuberculosis, brucellosis, or toxoplasmosis; with inflammatory syndromes such as acute glomerulonephritis or acute rheumatoid arthritis; and with liver failure or diabetic acidosis. Infants with Down syndrome may develop a transient myeloproliferative disorder (see Chapter 11 ) that is distinguishable by the marked leukoerythroblastosis with circulating blast cells. Leukemoid reactions have also been identified in neonates with thrombocytopenia-absent radius syndrome.
Clinical States Associated with Alterations of Eosinophil Numbers
Eosinophilia
A very broad range of conditions can elicit an eosinophilic response ( Box 22-8 ). Eosinophil stimulation most commonly occurs after repetitive or prolonged antigen exposure, especially when the antigens are deposited in the tissues and elicit hypersensitivity reactions, whether of the immediate (IgE-mediated) or delayed (T-lymphocyte–mediated) type. Stimulation of eosinophilia is T-lymphocyte–dependent and underlies the immune response to metazoan parasites.
|
|
Allergy is the most common cause of eosinophilia in children in the United States. Acute allergic reactions may cause leukemoid eosinophilic responses, with eosinophil counts exceeding 20,000 cells/µL, whereas chronic allergy is rarely associated with eosinophil counts of more than 2000 cells/µL. A variety of skin diseases have been associated with eosinophilia, the best documented being atopic dermatitis, eczema, pemphigus, acute urticaria, and toxic epidermal necrolysis. Drug reactions often elicit eosinophilia, including the potentially fatal drug rash with eosinophilia and systemic symptoms (DRESS) syndrome.
Outside the United States, and hence also in immigrants and returning travelers, parasitic infections are the most common causes of eosinophilia. Infestations by certain parasites, including helminths, induce greater degrees of eosinophilia than do protozoan infestations. Other parasites such as Giardia lamblia , Enterobius vermicularis, and Trichuris trichiura fail to elicit an eosinophilic response, probably because they remain localized to the intestinal tract and do not enter somatic tissue.
When parasites invade systemic organs, they may incite clinical symptoms and signs related to the involved organs, such as hepatomegaly and pulmonary infiltrates. Complications include seizures, encephalitis, myocarditis, retinal lesions (that are often difficult to distinguish from retinoblastoma), and skin nodules on the palms of the hands and soles of the feet. Leukocyte counts may exceed 100,000 cells/µL, with marked eosinophilia persisting from months to years after resolution of symptoms. Polyclonal hypergammaglobulinemia is common. Increased anti-A and anti-B titers are commonly observed because of cross-reactivity between red cell and parasitic antigens.
Eosinophilia is also observed in scarlet fever, CMV pneumonia of infancy, cat-scratch disease, infectious lymphocytosis, and occasionally, infectious mononucleosis. Many patients with acute pulmonary tuberculosis show decreased numbers of circulating eosinophils followed by an increase in eosinophil numbers during the convalescent phase, but rarely patients exhibit marked eosinophilia at diagnosis.
In general, fungal diseases do not cause eosinophilia, but important exceptions are allergic and nonallergic fungal sinusitis and rhinosinusitis, coccidioidomycosis, and allergic bronchopulmonary aspergillosis. The latter two diagnoses fit into the spectrum of pulmonary eosinophilic syndromes, which also include idiopathic eosinophilic pneumonia, Churg-Strauss syndrome (a form of vasculitis), tropical pulmonary eosinophilia (caused by microfilariae and other parasites), and drug- and radiation-induced pulmonary eosinophilia.
GI disorders may also be associated with eosinophilia. Primary eosinophil-mediated disorders usually involve the esophagus and stomach but can affect any or all segments of the GI tract. Eosinophilia can also be a secondary sign of food allergy. Inflammatory bowel disease and gluten enteropathy may involve large numbers of tissue eosinophils but show little or no elevation of blood eosinophil numbers.
Eosinophilia is a prominent feature of several immunodeficiency disorders, especially hyper-IgE syndrome (HIES; Job syndrome) (see later), Wiskott-Aldrich and Omenn syndromes, and hereditary angioedema. Approximately 10% of patients with rheumatoid arthritis develop mild eosinophilia during the course of their disease.
AD familial eosinophilia has been reported in several families in which individuals displayed marked eosinophilia, but few had pulmonary, cardiac, or neurologic involvement. The disorder has been mapped to chromosome 5q31-q33, a region that contains a cytokine cluster including genes encoding IL-3, IL-5, and GM-CSF. However, no mutations or functional polymorphisms have been identified within the promoter, exons, or introns of any of these genes, suggesting that the primary defect in familial eosinophilia is located in another gene in the cluster. The disorder can be distinguished from HES (see the following section) by the relative absence of eosinophil activation markers and rarity of end organ damage.
Marked eosinophilia can accompany or precede the diagnosis of malignancies including Hodgkin and non-Hodgkin lymphomas, acute lymphoblastic leukemia, and a variety of solid tumors. Excessive numbers of normal eosinophils may also be produced in response to production of cytokines, primarily IL-5, by abnormal (but only sometimes clonal) T cells with aberrant immunophenotypic markers. About one third of patients undergoing chronic hemodialysis develop blood eosinophilia without an apparent cause. Similarly, chronic peritoneal dialysis may cause an eosinophilic peritoneal effusion and occasionally an elevated number of eosinophils in the blood.
Eosinophilia can produce organ damage, particularly to the heart, lungs, and GI tract, by infiltration and deposition of toxic granule proteins. If treatment of the underlying infection or disease is not sufficient to reduce the eosinophil count, then steroid therapy generally produces a rapid decline in eosinophil production and circulating cell numbers. Steroid-sparing targeted therapies such as monoclonal antibodies directed at IL-5 or IL-5R (e.g., mepolizumab, reslizumab, and benralizumab) or at T cells (e.g., alemtuzumab) have shown efficacy, and a variety of additional biologic agents are in development.
Hypereosinophilic Syndrome
HES is defined as a persistent eosinophilia in patients that meet the following criteria: (1) absolute eosinophil count of 1500 cells/µL or more for longer than 6 months (or fatal termination within 6 months); (2) lack of other diagnoses to explain secondary eosinophilia; and (3) signs and symptoms of organ involvement by infiltrating eosinophils. Clinical manifestations result from tissue infiltration by eosinophils and the release of eosinophil granule products that cause tissue damage. Symptoms include nonspecific findings of fever, weight loss, and fatigue. Cardiac damage is the major cause of morbidity and mortality in HES and includes endocardial fibrosis and formation of mural thrombi with infiltrating eosinophils (Loeffler endocarditis). These findings can also be seen from eosinophilia of multiple other etiologies, including parasitic infection, drug reactions, or secondary to malignancies. Hepatosplenomegaly, pulmonary infiltrates, and skin involvement with urticarial or nodular lesions are common; neuropathies, encephalopathy, and CNS thromboembolism can also occur.
The recognition of specific gene rearrangements has provided a molecular basis for understanding the pathophysiology, developing classification systems, and guiding treatment. The most common involve the gene encoding the platelet-derived growth factor receptor α, PDGFRA , which forms fusion products that generate constitutively active tyrosine kinase molecules. The most common fusion partner is the Fip1-like 1 gene, FIP1L1 , resulting from an interstitial chromosome 4q12 deletion that is not cytogenetically detectable; however, multiple rarer fusions and cytogenetic changes have been reported. These rearrangements result in HES with a marked male predominance and a phenotype that includes splenomegaly, elevated serum vitamin B 12 and tryptase levels, and hypercellular bone marrow with fibrosis and increased eosinophils and mast cells. Other patients with clonal HES have a wide variety of rearrangements involving the PDGFRB gene, which encodes platelet-derived growth factor receptor β, and rare cases have been reported with rearrangements of FGFR1 , which encodes fibroblast growth factor receptor 1, and other genes. Importantly, patients with PDGFR fusion products are likely to respond to imatinib therapy, sometimes at dosages lower than those used for CML.
There is still no consensus on classification of HES, in part because of overlap between primary and secondary hypereosinophilia. The most general classification groups the syndromes as: (1) primary neoplastic HES with an underlying clonal abnormality; (2) secondary HES caused by an underlying neoplastic or nonneoplastic condition leading to nonclonal eosinophilia; and (3) idiopathic HES. The diagnosis of chronic eosinophilic leukemia is very difficult to distinguish from clonal HES, although the World Health Organization classification makes this distinction. The lymphoid form of secondary HES is a distinct variant caused by monoclonal expansion of T cells with an indolent course and rare evolution to T-cell lymphoma.
HES can generally be distinguished from the M4Eo variant of AML, which is characterized by myelomonocytic blasts with eosinophilia and inv(16) cytogenetics (see Chapter 11 ).
Eosinopenia
Eosinopenia is not uncommon but is seldom recognized clinically, because its precise diagnosis requires an absolute eosinophil count. Eosinopenia may be produced by at least two mechanisms: (1) primary elevation of adrenal corticosteroids or epinephrine; and (2) acute inflammation or stress, acting in part through secondary release of adrenal corticoids and/or epinephrine. The immediate eosinopenia after administration of glucocorticoids reflects the destabilization of mRNA encoding cytokines such as eotaxins and inhibition of the cytokine-dependent survival of eosinophils. Glucocorticoids also suppress the transcription of a number of genes involved in eosinophil production and trafficking, including IL-3, IL-4, IL-5, and GM-CSF. Acute inflammation is associated with alterations in eosinophil distribution and production; at least part of this response occurs independently of adrenal corticoid release.
Basophilia and Basophilopenia
Basophils are associated not only with hypersensitivity reactions of the immediate type but also have increasingly recognized roles in innate immunity, particularly to helminthes and other parasites. Basophil levels may be elevated in allergic responses; infections with helminthes, viruses, and mycobacteria; and chronic inflammatory diseases ( Box 22-9 ). Most importantly, basophil levels are increased in myeloproliferative disorders such as CML (see Chapter 11 ). Basophil counts exceeding 30% can occur during the course of CML; marked basophilia often heralds a poor prognosis, and the cells may be morphologically and functionally abnormal. Patients with marked basophilia may develop symptoms attributed to the release of biogenic amines or heparinlike material released from degranulated basophils and may benefit from the administration of antihistamines. Increased numbers of marrow basophils may occur in MDS and sideroblastic anemia. Peripheral blood or bone marrow basophilia may also accompany acute myeloid leukemias, usually in association with 6p or 12p chromosomal abnormalities, or juvenile myelomonocytic leukemia.
Mild Basophilia
Hypersensitivity reactions
Drug and food hypersensitivity
Urticaria
Infection
Helminths
Chickenpox
Influenza
Smallpox
Tuberculosis
Inflammation
Rheumatoid arthritis
Ulcerative colitis
Hypothyroidism (severe)
Marked Basophilia
Chronic myelogenous leukemia
Myelodysplastic syndrome
Acute myeloid leukemia (rare)
Basophilopenia occurs in conditions that are associated with eosinophilopenia, such as during acute infection or after the administration of glucocorticoids. Basophil counts are diminished in thyrotoxicosis and after treatment with thyroid hormones, and conversely they may be increased in myxedema.
Monocytosis and Monocytopenia
The normal absolute blood monocyte count varies with the age of the patient, and this variation must be taken into account when assessing monocytosis. During the first 2 weeks of life, the absolute monocyte count is more than 1000 cells/µL. With increasing age, there is a gradual decline in the monocyte count until it reaches a plateau of 400 cells/µL in adulthood. Monocytosis may therefore be defined as a total monocyte count of more than 500 cells/µL. Many clinical disorders give rise to monocytosis ( Box 22-10 ), most typically bacterial, protozoan, and rickettsial infections such as subacute bacterial endocarditis, tuberculosis, syphilis, Rocky Mountain spotted fever, and kala-azar. Monocytosis is a hallmark of juvenile myelomonocytic leukemia (see Chapter 11 ) and can also be observed in malignant disorders such as preleukemia, AML, CML, lymphomas, and advanced stage carcinomas. Approximately 25% of all patients with Hodgkin lymphoma have monocytosis, which does not correlate with prognosis. Monocytosis has also been noted in a wide variety of inflammatory and immune disorders including lupus, rheumatoid arthritis, sarcoidosis, and inflammatory bowel disease. Finally, as discussed, both relative and absolute monocytosis occurs in some forms of severe chronic neutropenia and in patients recovering from myelosuppressive chemotherapy.
Infection
Subacute bacterial endocarditis
Tuberculosis
Syphilis
Protozoal and rickettsial infections (e.g., Rocky Mountain spotted fever, kala-azar)
Malignancy
Preleukemia
Juvenile myelomonocytic leukemia
Acute myelogenous leukemia
Chronic myelogenous leukemia
Lymphoma (Hodgkin and non-Hodgkin)
Solid tumors (usually advanced carcinoma)
Rheumatologic Disorders
System lupus erythematosus
Rheumatoid arthritis
Sarcoidosis
Inflammatory bowel disease
Miscellaneous Disorders
Severe chronic neutropenia
Recovery from transient neutropenia
Postsplenectomy status
Alcoholic liver disease
Tetrachloroethane poisoning
Monocytopenia has been observed after glucocorticoid administration and in infections associated with endotoxemia. In the latter case, systemic activation of complement occurs with the deposition of C5a on the surface of monocytes, leading to their aggregation and clearance. Decreased monocyte superoxide production, chemotaxis, and microbial killing have been reported in Gaucher disease, possibly contributing to the risk of infections in this disease. The defects are reversed at least partially by enzyme replacement therapy.
Monocytopenia is a primary feature of the recently described MonoMAC syndrome. This pleiotropic disorder also includes, to varying degrees, B- and NK-cell deficiencies; mild neutropenia; susceptibility to severe and chronic nontuberculous mycobacterial, fungal, and viral infections; predisposition to MDS, myeloid leukemias, and autoimmunity; pulmonary alveolar proteinosis; and lymphedema. Deficient NK-cell function reflects the selective loss of CD56 bright cells. The syndrome includes phenotypes previously reported as Emberger syndrome (AD deafness, lymphedema, and MDS/AML) and NK-cell deficiency with severe herpes virus infections. MonoMAC syndrome derives from hypomorphic and regulatory mutations that diminish expression of the GATA-2 transcription factor, which regulates development and function in multilineage hematopoiesis and lymphangiogenesis, as well as leukemogenesis. The disorder has been treated successfully by HSCT.
Monocytopenia is a feature of another immunological disorder, so far unnamed, that is caused by mutations in the IRF8 gene and resulting in monocytopenia, absent or deficient dendritic cells, and immunodeficiency presenting as disseminated bacille Calmette-Guérin (BCG) disease.
Infantile (malignant) osteopetrosis is a hereditary disease of bone marrow replacement by bone that is caused by a failure of bone resorption and remodeling by osteoclasts, a form of specialized tissue macrophage. This disorder presents in infancy and is characterized by the progressive obliteration of the bone marrow space by bone, leading to progressive loss of marrow function, hepatosplenomegaly, growth retardation, and compression of cranial nerves. AR and less severe AD osteopetrosis are caused by mutations in any of multiple genes involved in receptor activator of nuclear factor κB and its ligand (RANK and RANKL) signaling, receptor-mediated endocytosis, vacuolar proton and chloride transport, G-protein degradation, or vesicular sorting.
Generation of superoxide by peripheral blood leukocytes is defective in some patients with AR (malignant) osteopetrosis and recombinant IFN-γ (also used for CGD; see later) has been shown to increase bone resorption and improve hematopoietic marrow function in children with infantile osteopetrosis. Infantile osteopetrosis is generally treated with bone marrow transplantation, Promising studies in mouse models suggest that RANKL administration or gene therapy might provide future therapeutic options.
Disorders of Granulocyte and Mononuclear Phagocyte Function
Abnormalities in one or more steps of phagocyte function—adhesion, chemotaxis, ingestion, degranulation, and oxidative metabolism—are causes of inherited and acquired clinical disorders that can be classified based on the function primarily affected. Consistent with the critical role of phagocyte function in host defense, patients afflicted with these disorders often suffer from recurrent, difficult-to-treat bacterial and fungal infections in skin or mucosa, lung, lymph nodes, or deep-tissue abscesses. Many of these disorders have characteristic clinical and microbiologic features related to the particular functional defect. Finally, inherited disorders involving cytokine or TLRs and their signal transduction are increasingly recognized, many of which affect phagocyte function and result in recurrent infections. These are described near the end of this chapter.
Of note, in clinical practice most physicians encounter a number of patients who suffer from recurrent bacterial infections. Whereas it is true that nearly all patients with well-characterized phagocyte abnormalities have recurrent infections, the converse is seldom the case. Most patients with impressive histories of persistent and repeated infections do not have identifiable qualitative or quantitative phagocyte abnormalities. Therefore the disorders described below account for only a fraction of patients with recurrent infections. This point will be discussed further in the final section describing the laboratory evaluation of phagocyte function.
Disorders of Adhesion
Leukocyte Adhesion Deficiency Type I
LAD I is a rare AR disorder in which phagocyte adhesion, chemotaxis, and ingestion of C3bi-opsonized microbes is impaired because of mutations in the gene for the β subunit of the β 2 integrins, CD18. As a result, the expression of β 2 integrins on leukocyte cell surfaces is reduced or absent. More than 100 patients with this disorder have been described in the literature. The hallmark of LAD I is the occurrence of repeated, often severe bacterial and fungal infections without the accumulation of pus despite a persistent granulocytosis ( Table 22-10 ). The clinical syndrome is heterogenous and is related to the severity of the reduction in β 2 -integrin expression. Most patients have a severe clinical phenotype, in which fewer than 0.3% of the normal amount of β 2 integrins are present, although a more moderate phenotype with levels of 2.5% to 11% of normal has been observed. Patients with the severe form of LAD I generally show signs in early infancy with neutrophilia, omphalitis, and delayed separation of the umbilical cord. Recurrent necrotic and indolent infections of skin, mucous membranes, and the GI tract are also seen, including perirectal abscesses, which often heal poorly. An aggressive form of gingivitis and periodontitis is characteristic, and ulcerative lesions of the tongue and pharynx can be seen. Otitis media and pneumonia are also often encountered. Most infections are caused by S. aureus and gram-negative enteric bacteria. Fungal infections also occur, particularly Candida albicans and Aspergillus species.
| Chronic Conditions * | Acute Infections | Infecting Organisms | Other |
|---|---|---|---|
|
|
| Delayed umbilical cord separation |
* Each list is arranged in approximate order of incidence based on reviews summarizing various series of patients with leukocyte adhesion deficiency.
The molecular basis for LAD I was first suggested by Crowley and colleagues, who found that neutrophils from a patient with this clinical syndrome lacked a high–molecular-weight membrane glycoprotein. Because the patient’s neutrophils neither adhered to plastic surfaces nor underwent an oxidative burst when exposed to serum-opsonized particles, it was hypothesized that the missing glycoprotein was responsible for both adhesion and cell-particle interactions. A similar glycoprotein was also found to be missing in several other patients, and that proved to be the α subunit (CD11b) of the Mac-1 β 2 integrin (CD11b/CD18). It was subsequently recognized that the levels of all leukocyte β 2 integrins (see Fig. 22-5 ) were absent or severely deficient in LAD I.
Deficient β 2 -integrin expression impairs a variety of leukocyte adhesion-dependent activities, with neutrophils being the most significantly affected. In addition to their role in mediating adhesive interactions during neutrophil emigration and phagocytosis, signaling through β 2 integrins potentiates virtually all functional responses of adherent neutrophils, including production of reactive oxidants and degranulation. Hence LAD I neutrophils have a profound defect in their ability to become fully activated.
A striking finding in LAD I is the failure of neutrophils to migrate to sites of inflammation. The initial phases of neutrophil adhesion to the endothelium (see Fig. 22-3 ), which is mediated by selectins and their sialylated counter receptors, is normal in LAD. However, because of deficient Mac-1 (CD11b/CD18) expression, LAD I neutrophils are neither able to attach firmly to the endothelium nor undergo transendothelial migration. Neutrophil intravascular survival is prolonged, presumably related to deficient adhesion and migration. In vitro assays of neutrophil adhesion to glass or to cultured endothelial cells and of neutrophil chemotaxis also exhibit marked abnormalities. An exception to these observations is neutrophil adhesion and emigration in the pulmonary capillary bed, which can be mediated by CD11/CD18-independent mechanisms. In autopsy tissue from a patient with severe LAD I, no neutrophils were observed in infected appendiceal and skin lesions, whereas many neutrophils were seen within the alveolar spaces. In contrast to neutrophils, other leukocytes (monocytes, eosinophils, and lymphocytes) express the β 1 integrin, VLA-4 and are able to utilize this adhesion molecule to emigrate into inflammatory sites throughout the body.
Another major defect in LAD I is the inability of neutrophils and monocytes to recognize microorganisms coated with the opsonic complement fragment C3bi, since Mac-1 is a major C3bi receptor. The binding of C3bi normally triggers neutrophil degranulation, phagocytosis, and activation of the respiratory burst, but these responses are diminished or absent in neutrophils from patients with LAD I. However, these responses can be at least partially activated in LAD I neutrophils by opsonins (e.g., IgG, C3b) that have different cell surface receptors or by soluble agonists that bypass Mac-1.
Defects in LAD I lymphocyte functions dependent on LFA-1 (CD11a/CD18) are often reported when assayed in vitro. These include proliferative responses to mitogens, NK-cell function, and lymphocyte-mediated killing. Nevertheless most patients with LAD I manifest few if any problems related to lymphocyte dysfunction in vivo. Cutaneous hypersensitivity reactions are normal, and patients are generally not unusually susceptible to viral infections, including varicella. However, one patient has died of an overwhelming respiratory infection with picornavirus, and one or more episodes of aseptic meningitis have been reported in three patients.
Molecular Basis of Leukocyte Adhesion Deficiency.
That LAD I involves a deficiency of all leukocyte β 2 integrins focused attention on the common β 2 chain (CD18) of this integrin family as the site of the molecular defect. This hypothesis proved to be correct. Expression of the leukocyte-integrin α subunits is normal in LAD I, but these are not transported to the cell surface because the β 2 chain is absent or contains mutations that disrupt β 2 structure or its interaction with the α subunit. More than 80 different LAD I-associated mutations in the β 2 gene IBTG2 , located on chromosome 21q22.3, are reported. A database is available at http://bioint.uta.fi . Many patients are compound heterozygotes for two different mutant alleles, whereas others are homozygous for a single mutant allele. About one half of patients with LAD I and characterized genetic defects have point mutations that result in single amino-acid substitutions in CD18 that almost invariably reside between amino acids 111 and 361 encompassed by exons 5 to 9. This protein domain is highly conserved among all β subunits, and appears to be important for interaction with the α subunit. In this LAD I subgroup, approximately one half exhibit a low level of CD11/CD18 cell-surface expression and moderate disease, with the remainder having absent expression and the severe phenotype. Other LAD I-associated mutations include mRNA-splicing defects as well as small deletions within CD18 coding sequences, resulting in a premature termination signal. Mutations resulting in nonfunctional but normally expressed β 2 occur, but very rarely.
There are several animal models of LAD I. These include the occurrence of a severe form of the disease in an Irish setter born of a mother-son mating and in Holstein cattle. In the latter case, affected calves could be traced to a common sire and were homozygous for a point amino-acid substitution in the conserved extracellular domain of CD18. Finally, CD18-deficient mice have produced by gene targeting.
Diagnosis and Treatment of LAD I.
The diagnosis of LAD I should be suspected in any infant or child who has unusually severe or recurrent infections and/or periodontitis accompanied by persistently elevated peripheral blood neutrophil counts, particularly if there is a paucity of neutrophils at affected sites and/or there is a delayed separation of the umbilical cord (see Table 22-10 ). However, a well infant with delayed separation of the umbilical cord and normal blood counts is very unlikely to have LAD I. Although the mean age of cord separation is reported to be 7 to 15 days, 10% of healthy infants can have cord separation at 3 weeks of age or later. A rare cause of recurrent, deep-seated tissue infections in association with neutrophilia and poor formation of pus is mutation of the Rac2 GTPase, which leads to neutrophil signaling defects affecting chemotaxis and adhesion (see “ Disorders of Chemotaxis ”).
The diagnosis of LAD I is established by flow cytometry to assess cell surface expression of any of the β 2 -integrin α (CD11) subunits or the shared CD18 subunit ( Fig. 22-14 ). Neutrophil Mac-1 deficiency is more dramatic if neutrophils are first stimulated, which normally upregulates Mac-1 cell-surface expression. Carriers of LAD I can generally be identified by flow cytometry, because they typically express approximately 50% of the normal level of β 2 integrins on leukocyte cell surfaces. Commercial genetic testing is also available.