Fig. 14.1
Mode of action of synthetic long peptide vaccination. The tumor-associated antigens are efficiently delivered by SLP easily obtaining access to DC in the vaccine injection area. Antigen-loaded DC migrate via afferent lymphatics to vaccine-draining lymph nodes to settle into para-cortical T cell areas, where antigen processing from ingested SLP continues, after which they engage in antigen presentation to CD4+ and CD8+ T cells. Activated T cells leave the lymph nodes via efferent lymphatic vessels ideally migrating to sites of tumor antigen expression in tumors. There a hostile environment is encountered, including hostile cellular elements such as myeloid-derived suppressor cells (MDSC) and Tregs as well as immunosuppressive chemokines and cytokines, expression of the enzyme IDO, and ligands for inhibitory T cell checkpoints like PD-1 and LAG-3. Together these conditions impair T cell migration, function, and expansion. (Adapted and updated from Melief et al. [6]
14.3 What Makes a Good Therapeutic Cancer Vaccine?
Recently we have reviewed the factors that determine success or failure of cancer vaccines [6, 7]. A successful cancer vaccine has to meet each of four requirements: (1) selection of the right tumor antigens, (2) choice of an efficient vaccine platform, (3) use of the proper vaccine adjuvant(s), and (4) use in combination with the proper co-treatment. The critical success factors and the corresponding choices to be made for success are listed in Table 14.1.
Table 14.1
Critical success factors in development of therapeutic cancer vaccines
Selection of the right tumor-associated antigens | E.g., viral targets, neo-antigens |
Avoid (central) tolerance | |
Choice of an efficient vaccine platform | Avoid antigenic competition: concentrated , defined and quantified pure cancer antigen source (DNA, RNA, SLP) |
Avoid inefficiency and undefined nature of protein/RNA cancer cell extracts | |
Exploit excellent processing by dendritic cells in vivo: durable CD4 and CD8 responses | |
Use of the proper built-in or added adjuvant(s) | Robust DC activating agents that strengthen and broaden T-cell response |
Th1 polarization | |
Use with proper combination treatment | Drive tumoricidal T cells and overcome hostile cancer micro-environment |
Chemotherapy, Checkpoint blockers, TNFR family agonists, gammaC cytokines, TGFβ antagonists |
With respect to the choice of the right antigens, cancer-associated antigens that have not been subject to central thymic tolerance-driving mechanisms are favored, in particular the antigens encoded by cancer viruses such as human papillomavirus (HPV), Epstein-Barr virus, human T-lymphotropic virus I (HTLV-I), hepatitis B virus (HBV), hepatitis C virus (HCV), Kaposi sarcoma virus (KSV), and Merkel cell carcinoma virus. The most important antigens encoded by these viruses have been reviewed in [6]. Together cancer viruses are the cause of approximately 20% of all cancer worldwide. An emerging and very important category of antigens expressed by an even larger proportion of human cancers are neo-antigens caused by mutations in cancer cells induced by, e.g., chemical carcinogens in cigarette smoke (lung cancers, H&N squamous cell cancers, bladder cancers), UV light (melanomas and basal cell carcinomas of the skin), or DNA repair deficiencies (e.g., microsatellite instability colorectal cancers). The importance of neo-antigens as highly effective targets for cancer-specific T cells follows from the marked correlation observed between clinical efficacy of checkpoint blocking monoclonal antibodies against CTLA-4 or PD-1 and the number of mutations in cancers of individual patients [8–10]. In one patient successful therapy with anti-CTLA-4 antibody was indeed associated with the expansion of a major tumoricidal Tc population directed against a UV-induced mutation [11]. In mouse models it was possible to predict and validate mutation-derived neo-antigens and vaccinate successfully with neo-antigen-containing SLP [12] [13, 14] or an RNA-based vaccine [15]. Similar efforts have been applied in a first proof of concept of immunogenicity of neo-antigens in the clinic with peptide-loaded DC [16]. Although the non-self-antigens of viruses and mutation-derived neo-epitopes are favored cancer vaccine ingredients, the T cell repertoire against many overexpressed antigens of the cancer-testis variety, against differentiation antigens, and against antigens such as survivin, Wilms tumor 1 (WT-1), wild-type or mutant p53, and mesothelin may be sufficient to induce robust T cell responses with antitumor activity (reviewed in [6, 7].
14.4 T Cell Epitope-Based Vaccination
Classical preventive vaccines consist of heat-killed or attenuated microorganisms that express properly folded proteins to induce protective antibodies against conformational antigens. Therapeutic vaccines, however, need to induce CD4 and CD8 T cell responses against defined T cell epitopes, presented by HLA class II or I molecules. The epitopes only assume the right conformation when bound to the HLA molecules. Peptide vaccines for induction of T cell responses thus consist of the exact length peptide constituting an epitope, or of a longer peptide (synthetic long peptide, SLP) that contains one or more epitopes, but needs further processing by antigen-presenting dendritic cells (DC) for proper binding of the precise epitope(s) into HLA molecules. Both exact fitting peptides (short peptides) and SLP (long peptides) have been used in therapeutic vaccines, but SLP are preferred for reasons outlined in the next paragraph. An advantage of epitope-based vaccines is that only amino acid sequences of epitope-rich regions of proteins need to be represented, and those segments of proteins that do not contain appreciable numbers of T cell epitopes can be omitted. Quite apart from containing immunologically silent sequences, proteins are also poorly processed by DC for presentation (so-called cross-presentation) to T cells, in particular CD8 T cells, whereas SLP are efficiently processed for presentation by HLA class II and class I, respectively, to CD4+ and CD8+ T cells [17]. In composing SLP vaccines, it is important to include epitopes capable of binding to the most common HLA class I and II molecules. To this end enough length of amino acid sequence needs to be represented in the peptide vaccine to harbor binding motifs for the most common HLA class I and II molecules, associated with the capacity to become efficiently processed by either the proteasome in the cytosol, followed by transport through the transporter of antigen processing (TAP) (HLA class I processing pathway) or cathepsins in the endosomes (HLA class II processing pathway) of DC [5].
14.5 Short Peptides Versus Long Peptides for Cancer Vaccines
Initial attempts with peptide vaccines involved single MHC class I binding epitopes + adjuvant in mice. Although such vaccinations showed antiviral or antitumor activity in several instances [18–20], reviewed in [21], such short peptide vaccines (<15 amino acids) are clearly suboptimal for a number of reasons. First, short peptides can directly bind exogenously to HLA class I molecules of all nucleated cells without antigen processing. Consequently, short vaccine peptides will end up at high concentrations in HLA class I molecules of nonprofessional antigen-presenting cells (APC) in the absence of co-stimulatory molecules, causing either short-lived Tc responses or even Tc tolerance [22–24]. A second disadvantage of exact class I binding short peptide vaccines is the absence of class II epitopes for CD4+ Th cell activation. Such Th cells are needed for optimal induction and maintenance of Tc effector responses ([25–29] and for generation of Tc memory cells [30–32]. Also, CD4+ T cells can themselves exert important antiviral or antitumor effector functions [26] [33]. Vaccination with short peptides formulated in incomplete Freund adjuvant (IFA) leads to weak or abortive T cell responses [22, 33–35], in part because of the previous problems, compounded by the fact that T cells elicited by short peptides in IFA travel to the vaccine site rather than to the tumor [34]. Vaccination with SLP (>20 amino acids long), but not short peptides in IFA or the related adjuvant Montanide ISA-51, in contrast, leads to sustained CD8+ T cell responses and robust antitumor immunity [22, 34, 36, 37]. Indeed vaccination with SLP in IFA or Montanide supports robust and sustained effector and memory T cell responses also in patients with premalignant lesions or cancer [38–43]. To some extent tolerance induction by short peptides can be overcome by vaccination with mature DC, loaded ex vivo with short peptides prior to injection [44, 45] or replacement of the Th signal with agonist antibody against CD40, with or without TLR3 ligand poly I:C [46, 47]. Upon subcutaneous injection of SLP, or in vitro, DC, but not other types of cells, are capable of effectively processing the SLP for effective MHC class I and II presentation [17, 48, 49]. In addition processing of SLP for MHC class I or II presentation is much more efficient than that of protein [17, 49]. SLP are capable of induction of both potent CD4+ and CD8+ responses in preclinical and clinical settings [36, 38, 39, 42, 50–52], whereas vaccination with intact proteins is associated with demonstrable CD4+ but not CD8+ responses [53, 54].
14.6 Alternative Vaccination Platforms
Delivery of concentrated antigen in a platform that allows efficient access to DC for HLA class I and class II presentation is important for any successful therapeutic vaccine aiming at an effective level of therapeutic T cells [6, 55]. Next to SLP, other powerful platforms are DNA or RNA vaccines. DNA vaccination is usually delivered with electroporation, as in two types of therapeutic vaccines against lesions caused by high-risk HPV16 [56, 57], and a successful RNA platform is delivery of tumor antigen-encoding RNA in cationic liposomes [15]. Viral vectors have also been used for therapeutic cancer vaccination. Viral vectors contain vector sequences that show antigenic competition with the inserted tumor antigens [58, 59], a problem that can be overcome to some degree by priming and boosting with two different viral vectors, the immune responses against which cross-react at the boost level against the inserted tumor antigen, but not at the vector level as in the PROSTVAC therapeutic vaccine directed against prostate-specific antigen (PSA) [60]. Interestingly, SLP can also be used to effectively boost CD4 and CD8 T cell immune responses in Rhesus macaques against conserved HIV antigens after priming with a replication competent poxvirus (NYVAC-C-KC) encoding conserved HIV antigen sequences [61].
14.7 Efficacy of SLP Vaccination Depends on Addition of a Strong Adjuvant
Although SLP enter easily into DC for processing into the HLA class I and II pathways, these DC need to be activated by a powerful adjuvant into mature DC that express not only the proper HLA-peptide complexes but also the necessary co-stimulatory molecules for induction of proper effector and memory CD4+ and CD8+ T cell responses [36]. Adjuvants that serve this purpose are poly I:CLC (TLR3 ligand), CpG (TLR9 ligand), incomplete Freund’s adjuvant-like Montanide ISA-51, or stimulator of IFN genes (STING) agonists (reviewed in [6]). The action of SLP vaccines can be further enhanced by attaching a TLR ligand such as CpG or Pam3Cys, a TLR 1/2 ligand, to the SLP by covalent coupling [62–64].
14.8 Clinical Efficacy of SLP Vaccines in Patients with Premalignant Disease
SLP vaccination appears to be very efficient to eradicate premalignant lesions caused by high-risk human papillomavirus type 16 (HPV16). HPV16 causes approximately 50% of cervical cancers and the large majority of HPV-positive head and neck cancers. Robust CD4+ and CD8+ T cell responses were induced in our studies of vulvar intraepithelial neoplasia, a premalignant disease of the vulva, caused by HPV16. In this case a vaccine containing 13 overlapping long peptides (25–35 amino acids long, together covering the entire amino acid sequence of 257 amino acids of the combined HPV16 E6 and E7 oncogenic proteins), was delivered subcutaneously as an emulsion in the mineral oil adjuvant Montanide ISA-51 at a dose of 300 μg per peptide. More than 50% of the patients benefited from partial or complete regression of lesions [38, 42]. Moreover we observed a highly significant correlation between the strength of the vaccine-induced T cell response and the clinical response [38, 42]. Recently, we reported similar results in twice the number of VIN patients [41]. Again a strong correlation between vaccine-induced T cell responses and clinical responses was observed, and those patients with histologically complete lesion regression had also lost demonstrable papillomavirus in the original skin lesions sites [41]. In patients with cervical cancer, the same vaccine was not as immunogenic, and clinical benefit could not be demonstrated in late-stage (recurrent, metastatic) cervical cancer patients [65], pointing toward an immunologically compromised situation in these patients and the need for additional combination therapy (see below).
14.9 Clinical Efficacy of Therapeutic SLP Vaccines in Cancer Patients Requires Combination Treatment That Overcomes the Hostility Toward T Cells Within Cancers
If the T cells that have responded to tumor-associated antigens do not succeed in eradicating cancer cells right away, a chronic situation may arise which has been well described by Schreiber and colleagues as an equilibrium between tumor cells and the immune system, manifesting itself as latency of tumors due to an ongoing T cell immune response [66]. Eventually tumors may escape from control by T cells through a variety of mechanisms (reviewed by [7, 66]). One of the least surprising conditions that allows tumors to escape is that the microenvironment in which tumors arise characteristically does not set the stage for robust effector T cell and memory T cell responses of the type seen in acute viral infections. Acute viruses incite a spate of immediate powerful danger signals generated by the virus, which strongly activate the innate and adaptive immune systems. Interestingly many cancers caused by oncogenic viruses start with long-term (many years!) persistent infection with a cancer virus such as human papillomavirus (HPV) or hepatitis B virus (HBV). Chronic persistent viruses employ many immune evasion and stealth mechanisms that undermine both innate and adaptive immunity to establish persistent infection (reviewed for HPV in [67]. Also the T cell host defense machinery adapts to chronic persistent infection by avoiding immunopathology through T cell checkpoint regulation [68–70] [7]. This of course has dire consequences if the persistent virus happens to have oncogenic properties. T cell immunogenic tumors with many mutations employ very similar mechanisms, also engaging in T cell checkpoint regulation and antigen loss [13, 71, 72]. Usually escape through toning down of the immune response is brought about by interaction between an ongoing T cell immune response and the cancer cells. For example, PD-L1 expression is induced on cancer cells by interferon γ released from antigen-specific tumor-infiltrating T cells (reviewed by [73]. Some of the most dangerous escape mechanisms from T cell immunity are complete loss of HLA class I expression from cancer cells by deletion of β2 microglobulin, an essential molecule for expression of HLA class I at the cell surface or loss of responsiveness of tumor cells to interferon γ [74]. Fortunately these instances are relatively rare. In fact in most cases countermeasures against immune escape are possible. For example, T cell–intrinsic inhibition of function can involve other mechanisms than PD-1-PD-L1 inhibition such as egr2-driven expression and inhibition by LAG3 and 4-1BB (CD137). If this is the case, rescue of tumoricidal T cell activity is possible by inhibition of LAG3 and application of agonistic antibody against 4–1 BB [75]. Also, in case of loss of an immuno-dominant neo-antigen, second in line neo-antigens in the hierarchy of immuno-dominance can take over, particularly following vaccination against these subdominant neo-antigens or following PD-1 checkpoint blocking [13]. This last example illustrates that spontaneous generation of sufficient T cells to tumor-associated neo-antigens may be lacking in many potentially immunogenic tumors that are not spontaneously rejected. Likewise an HPV16 E6/E7-positive tumor in mice does not spontaneously generate TIL that infiltrate the tumor and eradicate it, even though the viral antigens are expressed by the tumor. However a combination of cisplatin or carboplatin and paclitaxel chemotherapy and HPV-specific SLP vaccination is able to generate tumor-infiltrating HPV-specific TIL that completely eradicate established tumors [43, 76]. The mechanisms by which this type of chemo-immunotherapy operates are manifold and include immunogenic cell death causing DC activation (reviewed in [77]), synergistic apoptosis by TNF-α, released in the cancer microenvironment from vaccine-induced TIL in combination with cisplatin [76, 78], and depletion of immunosuppressive mononuclear myeloid cells that are present in excessive numbers in the blood and probably the cancers of many late-stage cancer patients [43]. An attractive choice for combination with therapeutic vaccines is the combination with checkpoint blockers, in particular anti-PD-1. Synergy between therapeutic vaccines and anti-PD-1 checkpoint blocking has been noted in a number of preclinical models [52, 79, 80]. Vaccination with autologous DC loaded with melanoma-associated antigens may act synergistically with CTLA-4 blocker ipilimumab in mediating clinical responses in late-stage melanoma patients [81], but this observation needs to be confirmed in a randomized study. Synergy has also been reported between therapeutic vaccines and one of the following agonistic monoclonal antibodies: anti-CD27, anti-CD40, anti-CD134 (anti-OX40), or anti-CD-137 (anti4-1BB) (reviewed in [6]). In addition inhibitory monoclonal antibodies have been used against the cytokines IL-10, TGFβ, and IL-6 (reviewed in [6]). An overview of the different attractive combination therapies with therapeutic vaccines is shown in Fig. 14.2.
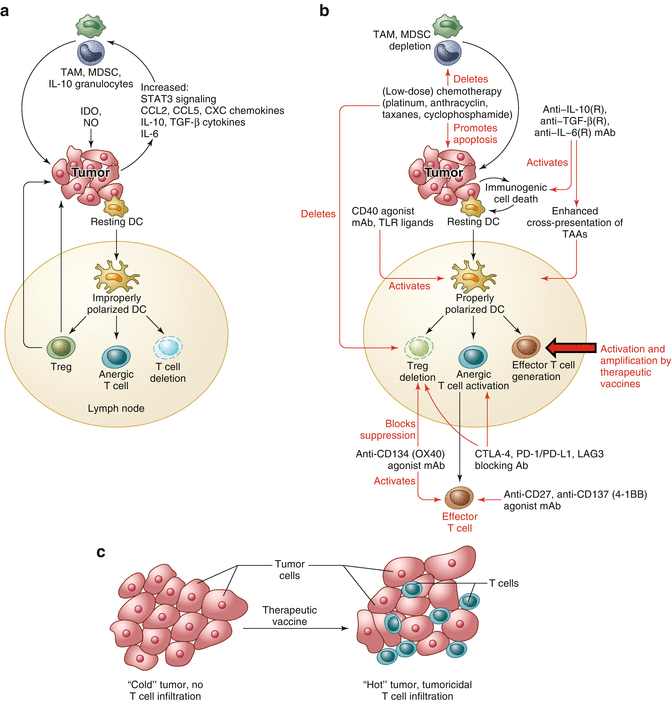
Fig. 14.2
Vaccines synergize with immunomodulators to yield an attractive combination treatment of cancer. (a) Hostile elements for T cells in the cancer microenvironment include tumor-associated macrophages (TAM), myeloid-derived suppressor cells (MDSC), and IL-10-producing granulocytes. In addition a number of immunosuppressive cytokines and chemokines are released in this milieu. Nitric oxide and the enzyme IDO may further compound the immunosuppressive situation. (b) Vaccines will expand the low numbers of spontaneous anticancer T cells and generate de novo T cells against new antigens, increasing the chances of success of the therapy compared to monotherapy with immunomodulators. (c) SLP vaccines can thereby turn “cold” tumors (no T cell infiltrate) into “hot” tumors (T cell infiltrate). The immunomodulators (shown in red in (b)) can repeal the immunosuppressive cancer microenvironment in a number of ways, according to each cancer patient’s needs. Thereby vaccines and immunomodulators synergize for effective cancer therapies
Recently we showed in a pilot trial in patients with late-stage (recurrent or metastatic) cervical cancer that much better HPV16-specific T cell responses could be induced by SLP vaccination in Montanide adjuvant by combination with standard of care chemotherapy consisting of carboplatin and paclitaxel [43]. The chemotherapy acted as an immunomodulator in the sense that T cell numbers and function were not affected by this chemotherapy combination, but the suppressive mononuclear myeloid cell levels found in all patients declined to those of healthy donors onward from 2 weeks into the second of six cycles of chemotherapy. A single vaccine dose given at that time point led to a much higher and broader T cell response against HPV16 E6/E7 [43] than observed in the previous trial without timed chemotherapy [65]. Based on these observations, we have now concluded a much larger clinical trial in 70 patients with late-stage (recurrent or metastatic) HPV16+ cervical cancer. Interestingly, in this trial the first of three vaccine doses with the HPV16 SLP vaccine, called ISA101, was given again at the nadir of the mononuclear myeloid cell count in PBMC (i.e., 2 weeks after the second cycle of chemotherapy), and a highly significant correlation was subsequently observed between the strength of the vaccine-induced HPV16-specific T cell response and overall survival of the patients (unpublished observations).
14.10 Perspectives
Cancer vaccines have only recently shown clinical efficacy for the treatment of premalignant disease [38, 41, 42, 56, 57] and prostate cancer ([82]) as monotherapy. Combination treatments with various checkpoint blocking or agonistic monoclonal antibodies are highly promising for the treatment of cancer, provided the right cancer target antigens are chosen in combination with efficient vaccine platforms and proper adjuvants. The best target antigens are clearly viral antigens on virus-induced cancers or neo-antigens on mutagen-induced cancers, such as those associated with smoking or UV light exposure. Overexpressed antigens or differentiation antigens may also be chosen, provided an appreciable repertoire of CD4+ and CD8+ T cells against the antigen of choice is demonstrable. Cancer vaccines are highly attractive in patients in whom the natural T cell response has fallen short, resulting in so-called “cold” tumors, which lack spontaneous infiltration with T cells. In many of these patients, therapeutic vaccination may cause sufficient numbers of vaccine-induced tumor-specific T cells to infiltrate into the tumors and turn them into “hot” tumors. This alone is unlikely to cause robust clinical responses, but co-treatment with a number of other cancer drugs, which each by themselves are also insufficiently effective, may reap a rich harvest of more effective combination treatments with relatively few side effects. One of the most attractive of these combinations is combination of therapeutic SLP vaccination with chemotherapy or with anti-PD-1 and/or CTLA-4 checkpoint blockers. SLP vaccines are particularly attractive for vaccination against neo-antigens created by point mutations in cancers, because of the relative ease of production under GMP conditions.
References
1.
2.
3.
Unanue ER. The regulation of lymphocyte functions by the macrophage. Immunol Rev. 1978;40:227–55.CrossrefPubMed
Related posts:
 Purinergic Receptors: Novel Targets for Cancer Immunotherapy
Purinergic Receptors: Novel Targets for Cancer Immunotherapy
 Current Status of Immuno-Oncology in Hematologic Cancers
Current Status of Immuno-Oncology in Hematologic Cancers
 Synergy Between Radiotherapy and Immunotherapy
Synergy Between Radiotherapy and Immunotherapy
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


