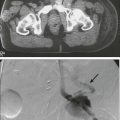
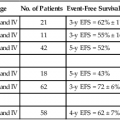
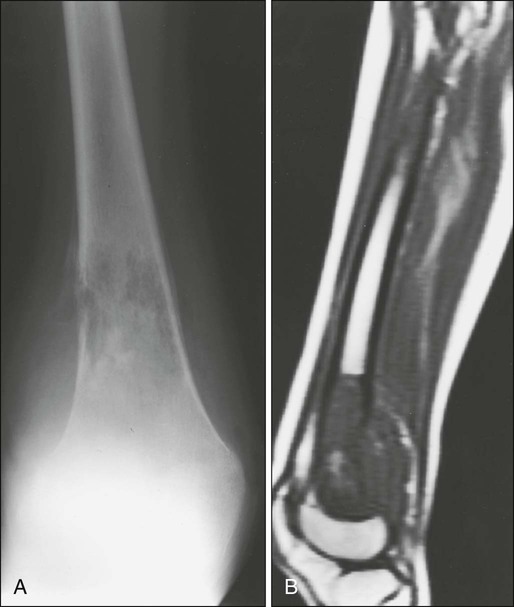
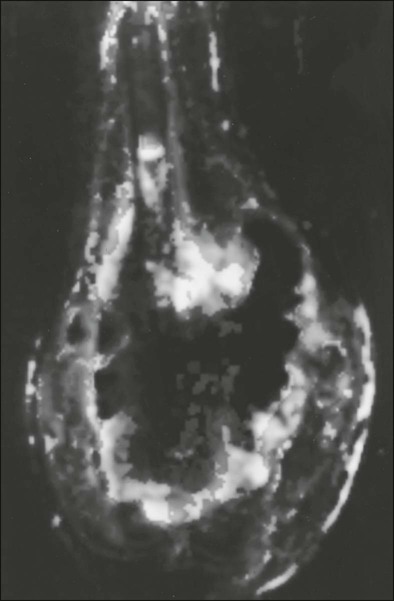
95 Jeffrey S. Dome, Carlos Rodriguez-Galindo, Sheri L. Spunt and Victor M. Santana • The incidence of osteosarcoma is four new cases per 1 million population per year among children younger than 15 years of age. • Osteosarcoma is the most common bone tumor in children and adolescents. • Other lytic bone lesions, including eosinophilic granuloma and giant cell tumor, must be excluded. • At histopathological examination, tumors must be differentiated from fibrosarcoma and chondrosarcoma. • Staging evaluation includes a complete history and physical examination, complete blood cell count (CBC), serum chemistry analysis (including determination of alkaline phosphatase level), and imaging studies of the primary tumor and chest. • The primary tumor is managed surgically with a limb-sparing operation or amputation. • Adjuvant chemotherapy incorporating high-dose methotrexate is used preoperatively for presumed micrometastatic disease; response will guide further therapy. • Methotrexate responders have a more than 80% chance of cure. • Primary pulmonary metastatic disease is managed surgically, although adjuvant chemotherapy may reduce the extent of resection. • Pulmonary metastatic disease is managed surgically. • Postthoracotomy adjuvant chemotherapy is of unproven benefit. • Local recurrence is managed surgically. • Further adjuvant chemotherapy with non–cross-resistant agents is of uncertain benefit. • The incidence of Ewing sarcoma family tumors is 2.8 new cases per 1 million population per year among children younger than 15 years of age. • Ewing sarcoma is the second most common bone tumor among children and adolescents. • Osteomyelitis must be ruled out, especially when the patient has a fever. • Lesions to be excluded include benign tumors of bone that manifest as lytic lesions (e.g., eosinophilic granuloma, giant cell tumor), malignant tumors (e.g., osteosarcoma, primary lymphoma of bone), and metastatic lesions from a nonosseous tumor (e.g., neuroblastoma). • Staging evaluation includes a complete history and physical examination, CBC, serum chemistry analysis (including determination of lactate dehydrogenase level), bone marrow biopsy, and imaging studies of the primary tumor, bones, and chest. • The primary tumor should always be treated with multimodality therapy consisting of chemotherapy, radiation therapy, surgery, or a combination of these treatments. • Specific local treatment depends on the primary site. Surgical extirpation may be considered with tumors in expendable bones (proximal part of the fibula, rib, clavicle, iliac wing). • Unresectable tumors generally necessitate a combined approach of chemotherapy, radiation therapy, and surgery. • Localized disease is curable with combined therapy in more than 70% of cases. Metastatic disease is curable in 30% to 40% of cases. • Effective second-line therapy has not been established. • Local recurrence may be amenable to surgical extirpation. • Among white children younger than 15 years of age, 10.5 new cases occur per 1 million population per year. Among black children, the incidence is 8.8 new cases per 1 million population per year. • Neuroblastoma is the most common extracranial solid tumor in children. • Disseminated bone disease can resemble systemic infection, inflammatory disease, osteomyelitis, or rheumatoid arthritis. • Paraneoplastic syndromes associated with neuroblastoma (vasoactive intestinal peptide syndrome, opsoclonus-myoclonus-ataxia syndrome) must be differentiated from primary inflammatory bowel disease and neurologic disease. • Neuroblastoma must be differentiated from other small, round, blue cell neoplasms of childhood (e.g., Ewing sarcoma, primitive neuroectodermal tumor, non-Hodgkin lymphoma, undifferentiated soft-tissue sarcoma). • In as many as 10% of tumors, catecholamines are not produced. In 1%, the absence of an obvious primary lesion confounds the diagnosis. • Staging evaluation includes a complete history and physical examination, CBC, serum chemistry analysis (including determination of lactate dehydrogenase level), quantitative urine catecholamines, bone marrow examination, radionuclide scintigraphy, and imaging studies of the primary tumor and chest. • Specific treatment depends on stage of disease, age of the patient, and biological features of the tumor. • With few exceptions (completely resected primary tumor, localized tumor with/without complete excision and favorable tumor biological features, infants with stable stage 4S disease), multiple-agent chemotherapy is the backbone of multimodality treatment. • Survival depends on stage and biological features of the tumor (e.g., histopathological subtype, MYCN gene amplification). • The overall survival rate for asymptomatic patients with favorable biology stage 1, 2a, or 2b tumors is greater than 90%. • Overall survival among patients with advanced-stage 3 or 4 with MYCN gene amplification disease is poor. Less than one-third of children with high risk disease are long-term survivors. • No second-line therapy has proved to increase survival. • The incidence of Wilms tumor is eight cases per 1 million population per year among children younger than 15 years of age. • Mean age at diagnosis is 44 months for unilateral tumors and 31 months for bilateral tumors. • Familial cases account for 1.5% of cases of Wilms tumor. • Associated with Wilms tumor is a syndrome comprising aniridia, genitourinary anomalies, and mental retardation (WAGR syndrome); Denys-Drash syndrome; and Beckwith-Wiedemann syndrome. • No firmly established environmental factors have been identified. • The main histologic subtypes are favorable and anaplastic. • Major implicated genes and loci include WT1 (11p13), IGF2 (11p15), FAM123B/WTX (Xq11), and CTNNB1/β-catenin (3p22). • Mutations of the p53 gene (TP53) are associated with anaplastic tumors. • Loss of heterozygosity (LOH) at chromosomes 1p and 16q and gain of chromosome 1q are associated with adverse prognosis. • An asymptomatic abdominal mass is found in most patients. • Other characteristic features include abdominal pain, hematuria, hypertension, and congenital anomalies (genitourinary malformations, aniridia, and hemihypertrophy). • Neuroblastoma is the main consideration in the differential diagnosis. • Other tumors to be excluded are renal neoplasms (clear cell sarcoma, rhabdoid tumor, congenital mesoblastic nephroma, renal cell carcinoma). • Benign renal processes (nephrogenic rests, multicystic or polycystic kidneys, hydronephrosis, renal carbuncles, hemorrhage) also should be ruled out. • Evaluation begins with complete history and physical examination, with careful attention to blood pressure and assessment for associated congenital anomalies. • Other components of the staging evaluation include CBC, serum chemistry analysis, urinalysis, abdominal ultrasonography, abdominal/pelvic computed tomography (CT) or magnetic resonance imaging (MRI), and chest CT. • Surgery: Surgical resection of the primary tumor usually precedes chemotherapy in the approach taken by the Children’s Oncology Group (COG). Presurgical chemotherapy is used in the approach taken by the International Society of Pediatric Oncology (SIOP). • Chemotherapy: Agents used depend on disease stage and favorable versus anaplastic histology: stages I and II, favorable histology—vincristine, actinomycin D; stages III and IV, favorable histology, and stage I, anaplastic histology—vincristine, doxorubicin, actinomycin D; stages II through IV, anaplastic histology—vincristine, cyclophosphamide, doxorubicin, carboplatin, etoposide. • Radiation therapy: Radiation therapy is included in management for stages III and IV, favorable histology, and stages II through IV, anaplastic histology. • Recurrent disease is effectively managed with radiation therapy and chemotherapy with agents not used for initial treatment. • Patients who initially received aggressive treatment may respond to cyclophosphamide or ifosfamide-, carboplatin-, and etoposide-based regimens. • Renal failure is seen in less than 1% of patients with unilateral tumors and 12% in patients with bilateral tumors. • Congestive heart failure may occur in patients who receive doxorubicin (occurs in 4.4% of patients). • Pregnancy-related complications in adulthood have occurred in girls who receive flank irradiation. • A second malignant neoplasm also may develop (6.7% cumulative incidence of solid malignancies at age 40 years). • With favorable histology, outcomes include 85% 4-year relapse-free survival rate and 90% 4-year overall survival rate. • With anaplastic histology, outcomes include 50% 4-year relapse-free survival rate and 50% 4-year overall survival rate. • The incidence of renal cell carcinoma (RCC) is four cases per 10 million population per year among children younger than 20 years of age. • Mean age at diagnosis in pediatric series is 9 years. • Other renal tumors of childhood, primarily Wilms tumor, should be ruled out. • Staging evaluation includes a complete history and physical examination, CBC, serum chemistry analysis, and imaging studies of the abdomen, pelvis, and chest. • Nephrectomy of the involved kidney constitutes definitive therapy for primary tumors. • For metastatic disease, enrollment in clinical studies of investigational agents is recommended. Tyrosine kinase inhibitors have shown benefit for translocation renal cell carcinoma. • Among children younger than 20 years, the incidence of rhabdomyosarcoma is 4.7 new cases per 1 million population per year. • Rhabdomyosarcoma is the most common soft-tissue sarcoma among children and adolescents. • Other benign and malignant soft-tissue tumors must be excluded. • At pathological examination, rhabdomyosarcoma must be differentiated from the other small, round, blue cell tumors of childhood (e.g., Ewing sarcoma, neuroblastoma, non-Hodgkin lymphoma). • Staging evaluation includes a thorough history and physical examination; CBC; serum chemistry analysis; imaging studies of the primary tumor, regional lymph nodes, lungs, and bones; and bone marrow examination. • Primary therapy is chemotherapy with surgery, radiation therapy, or both. Specific treatment depends on age of the patient, tumor primary site and histologic subtype, and extent of disease. • Mutilating surgery usually can be avoided because the tumor is sensitive to both chemotherapy and radiation therapy. • The overall survival rate exceeds 70%, but survival will depend on extent of disease. More than 90% of patients with localized, resectable, favorable histology tumors survive, but less than 20% of patients with metastatic disease survive. • Cure rarely is possible except for patients with botryoid tumors and those with embryonal tumors whose tumor arose in a favorable site and was completely resected at initial diagnosis. Nonrhabdomyosarcoma Soft-Tissue Sarcoma • Among persons younger than 20 years, 7.7 new cases occur per 1 million population per year. • Peaks in incidence occur among infants and among children older than 10 years of age. • Other tumors to be ruled out include benign soft-tissue tumors, rhabdomyosarcoma, and extraosseous Ewing sarcoma. • Staging evaluation includes a thorough history and physical examination and imaging studies of the primary tumor and lungs. • Imaging of regional lymph nodes, liver, bones, and brain is indicated in some clinical settings. • Surgical excision with or without radiation therapy is indicated for patients with resectable tumors. • Chemotherapy may provide some benefit for patients with high-grade tumors larger than 5 cm in diameter and for those with unresectable or metastatic tumors. • Survival depends on the size and grade of the tumor and extent of disease. Patients with localized tumors 5 cm or less in diameter or localized low-grade tumors larger than 5 cm in diameter that can be resected have a survival rate exceeding 85%. Approximately 50% of patients with high-grade tumors larger than 5 cm or with unresectable disease survive. The survival rate is approximately 10% among patients with metastatic disease. • Cure generally is possible only for patients with local recurrence amenable to surgical extirpation and for those with distant recurrence of low-grade tumor that proves to be surgically resectable. • Among children younger than 5 years of age,11 new cases occur per 1 million population per year. • The two clinical forms of retinoblastoma are as follows: (1) Hereditary, bilateral or multifocal (40% of cases)—This form, characterized by germline mutations of RB1, may be inherited from an affected survivor or a silent carrier parent or may be the result of a new germline mutation. (2) Nonhereditary, unilateral or unifocal (60% of cases)—15% of unilateral cases represent germline mutations. Clinical Manifestations and Differential Diagnosis • Clinical manifestations: Leukocoria is seen in more than 50% of cases, strabismus in 20% to 25%. • Differential diagnosis: Coats disease, retinopathy of prematurity, persistent hyperplastic primary vitreous, Toxocara uveitis, and toxoplasmosis need to be ruled out. • Approach to staging depends on tumor size and on the presence or absence of intraocular and extraocular extension. • Indirect ophthalmoscopic examination of both eyes with the patient under general anesthesia is essential. • Imaging studies helpful in staging include ultrasonography, orbital and cerebral CT, and MRI. • Bone marrow biopsy and cerebrospinal fluid examination are reserved for patients with extraocular disease, optic nerve involvement, or choroid invasion. • Treatment must be individualized and depends on laterality, potential for vision, and tumor extent. • Enucleation is reserved for cases in which no potential for useful vision remains. Cryotherapy and photocoagulation are useful for management of small primary or recurrent tumors. • Radiation therapy is the treatment modality of choice for controlling local disease and preserving vision with larger tumors. • Use of chemotherapy is restricted to patients with advanced intraocular disease or with extraocular disease. • Among children younger than 15 years of age, 2.2 new cases occur per 1 million population per year. • Mean age at diagnosis is 18 months. • The tumor is associated with familial adenomatous polyposis and Beckwith-Wiedemann syndrome. • An association with very low birth weight has been found. • The major histologic subtypes are fetal, embryonal, macrotrabecular, and small cell undifferentiated. • The tumor is associated with mutations of the APC and β-catenin genes and loss of heterozygosity at 11p15, the locus of the IGF2 gene. • An asymptomatic abdominal mass is present in most patients. • Other features may include anorexia, weight loss, vomiting, and precocious puberty (seen in 2% of cases). • Other malignant tumors to be excluded include hepatocellular carcinoma, embryonal sarcoma, rhabdomyosarcoma, angiosarcoma, and teratoma. • Benign tumors to be ruled out include hemangioma, hemangioendothelioma, hamartoma, and adenoma. • Staging evaluation includes a complete history and physical examination, with careful assessment for any congenital abnormalities or signs of precocious puberty. • Laboratory tests typically include CBC, serum chemical analysis, and α-fetoprotein and α-human chorionic gonadotropin assays. • Diagnostic imaging may include CT or MRI of the abdomen and chest CT. • Cure is possible only when complete surgical excision is performed. If complete excision is not feasible, liver transplantation should be considered. • Adjuvant chemotherapy (cisplatin-based, in conjunction with doxorubicin or 5-fluorouracil) is useful preoperatively for achieving resectability and postoperatively for preventing distant metastasis. • Radiation therapy has a limited and ill-defined role. • Recurrent disease confers a poor prognosis, but repeated resection of local and metastatic recurrences has lengthened the survival period. • Hearing loss and nephrotoxicity (related to cisplatin), cardiac toxicity (related to doxorubicin), and development of second malignant neoplasms may occur. • With localized disease, outcomes include a 60% to 70% 5-year relapse-free survival rate and a 70% to 80% 5-year overall survival rate. • With metastatic disease, outcomes include a 25% to 30% 5-year relapse-free survival rate and a 50% to 60% 5-year overall survival rate. • The incidence of adrenocortical carcinoma is two to three new cases per 10 million population per year in the United States. • A 10- to 15-fold higher incidence has been observed in southern Brazil. • Associated with germline TP53 mutations and Li-Fraumeni syndrome. • Other conditions associated with pathological androgen or cortisol production, such as Cushing syndrome or ovarian or testicular tumors, should be ruled out. • Evaluation for staging includes imaging studies of the abdomen, pelvis, and chest; skeletal scintigraphy; and measurement of blood and urine concentrations of adrenocortical hormones. • Complete surgical removal of the tumor is indicated. • Mitotane-based therapy or chemotherapy with cisplatin also is given. • Recurrent disease can be managed with further surgery, or experimental agents can be tried. Osteosarcoma, a malignant neoplasm derived from primitive mesenchymal cells and characterized by the presence of osteoid-producing spindle cell stroma, is the most common malignant bone tumor in the pediatric age group.1 Osteosarcoma ranks tenth among all newly reported pediatric cancers in the United States, accounting for 2.6% of all neoplasms in children. The estimated annual incidence is 3.9 cases per 1 million population among white children and 4.5 per 1 million population among African American children.2 Most osteosarcomas occur during the first 2 decades of life, a period characterized by rapid skeletal growth. Boys are affected more commonly than girls. Several observations support the association between skeletal growth velocity and osteosarcoma. First, patients with osteosarcoma tend to be taller than their counterparts without this disease. Second, osteosarcoma develops at an earlier age in female patients than in male patients, perhaps because of differences in the timing of onset of puberty and the growth spurt.3 Unlike osteosarcoma in adults, in whom more than 25% of tumors are associated with preexisting pathological osseous conditions such as Paget disease or fibrous dysplasia, most pediatric osteosarcomas arise spontaneously in areas of bone without any abnormality.1 Irradiation is the best-characterized etiologic factor contributing to the development of secondary osteosarcoma. In a study involving 91 patients with second malignant bone sarcomas, osteosarcoma accounted for 72 cases, 52 (72%) of these tumors arising within previously irradiated fields.4 The median time for development of the secondary tumor was 9.6 years after irradiation. Osteosarcoma as a second malignancy is often associated with retinoblastoma; osteosarcoma is the most common malignancy in survivors of retinoblastoma, both in the irradiated and the nonirradiated areas, and it accounts for 25% to 40% of all second neoplasms in this population.5,6 One half to two-thirds of osteosarcomas occur in the irradiated fields of the skull and face, one-third of tumors develop in the extremities, and less than 10% occur in the trunk. Osteosarcoma is also a very frequent malignancy in individuals with germline TP53 mutations (Li-Fraumeni syndrome)7 and in patients with REC helicase-associated disorders (Rothmund-Thomson, Werner, and Bloom syndromes.8 Consistent with the association of osteosarcoma with retinoblastoma survivors and Li-Fraumeni syndromes, alterations in components of the cell-cycle control system appear to characterize the ontogeny of osteosarcoma. Studies of the retinoblastoma gene (RB1) have shown that alterations affect the RB1 gene in as many as 80% of cases and that other events, such as CDK4 alterations, also may result in RB1 inactivation.9,10 Genetically engineered mice based on osteoblast-restricted deletion of p53 and pRb (retinoblastoma gene product) develop short-latency high-grade osteosarcoma that reproduces many of the defining features of human osteosarcoma, including cytogenetic complexity and comparable gene expression signatures, histology, and metastatic behavior.11 Other genetic contributions to the pathogenesis of osteosarcoma are less known; multiple cytogenetic abnormalities have been described, suggesting the presence of additional gene pathways involved in the multistep process of tumor development and progression.12 The peak incidence of osteosarcoma coincides with the adolescent growth spurt. This finding has led to the hypothesis that the altered hormonal milieu typical of adolescence may play a role in development of osteosarcoma. It is therefore possible that the insulin-like growth factor–1 (IGF1)—insulin-like growth factor–1 receptor (IGF1R) axis may be involved in the unregulated proliferation of osteoblasts that occurs in osteosarcoma. IGF1 functions as a mitogen in human and mouse osteosarcoma cells, and osteosarcoma cell lines depend on IGF1 for in vitro growth.13 Although the levels of IGF1 and its insulin-like growth factor binding protein–3 (IGFBP3) are not elevated in patients with osteosarcoma, other components of the IGF1 signaling pathway may be involved in the development and progression of osteosarcoma.14 Osteosarcoma is characterized by the presence of spindle cell stroma that produces osteoid. Conventional osteosarcoma can be subdivided histologically into three major groups depending on the predominant cell type. Approximately 50% of tumors are categorized as osteoblastic, because the predominant extracellular element is osteoid, whereas 25% are chondroblastic, with a prominent cartilaginous component. Approximately 25% have a herringbone pattern similar to that observed in fibrosarcoma, and are therefore called fibroblastic. No significant differences in overall outcome are apparent among these three histologic subtypes.1 In one report, however, patients with fibroblastic histology showed better histologic response and better overall outcome than were observed for patients with osteoblastic or chondroblastic histology.15 Pain is the most common symptom in children and adolescents with osteosarcoma.1 Onset of pain often is insidious, and the pain usually involves the area affected by tumor. Severe pain of sudden onset commonly is associated with pathological fracture. Swelling around the affected bone is the second most common clinical finding. The tumor may be easily palpable when located in areas such as the anterior surface of the femur but may manifest only as leg edema when occurring in difficult-to-appreciate areas such as the popliteal fossa. A painful limp that increases with weight bearing is the third most common symptom. Systemic signs and symptoms such as fever and weight loss are uncommon. Osteosarcoma most commonly involves the long bones, most tumors occurring around the knee. The most frequent sites of involvement are the distal part of the femur, the proximal portion of the tibia, and the proximal part of the humerus. The axial skeleton, including the pelvis, is rarely affected in children (fewer than 10% of cases) but more frequently is involved in patients older than 60 years.1,16 Overt macroscopic metastatic disease occurs in 20% of cases and carries a grave prognosis.17 Laboratory evaluation often is unrevealing. Elevations of serum lactate dehydrogenase (LDH) and alkaline phosphatase levels are the most common laboratory abnormalities. The latter appears to correlate with osteoblastic activity and thus has proved useful in monitoring response to therapy.18 Radiologic evaluation of a patient with osteosarcoma must include assessment of the primary site as well as a search for distant metastatic lesions.19 Plain radiography is the most effective method of detection of bone tumors. The main limitation of plain radiography is accurate delineation of local tumor extent. Characteristic radiologic findings in osteosarcoma commonly include a metaphyseal permeative lesion with periosteal new bone formation and destruction of preexisting cortical bone. A soft-tissue mass is present in more than 90% of cases. Other radiologic signs commonly associated with osteosarcoma include cumulus cloud-like density and the presence of Codman’s triangle (Fig. 95-1A). A baseline chest radiograph should be obtained to search for distant metastatic lesions. Angiography usually is reserved for patients who receive intraarterial chemotherapy or for those who need optimal vessel visualization before limb salvage. Computed tomography (CT) of the primary tumor is accurate in assessment of degree of tumor calcification and ossification, which are important in assessment of response to therapy. Chest CT always should be performed at the time of diagnosis for documentation of metastatic disease. Findings by magnetic resonance imaging (MRI) offer the best estimate of intramedullary tumor extension, joint and vascular involvement, detection of “skip” metastatic lesions, and delineation of the soft-tissue component (see Fig. 95-1B). The blood supply and vascularity of the tumor can be better appreciated with administration of gadopentetate dimeglumine, or gadolinium-diethylenetriamine pentaacetic acid (Gd-DTPA), a paramagnetic contrast material. MRI is helpful in assessing response to chemotherapy, as evidenced by changes in signal intensity on T2-weighted images and alterations in the enhancement of tumor tissue after administration of Gd-DTPA (Fig. 95-2). Dynamic contrast-enhanced MRI (DEMRI) is a valuable method for assessing microcirculation in osteosarcoma.20 DEMRI can be used to evaluate changes in regional contrast access during chemotherapy. The findings appear to correlate accurately with histologic response and outcome, allowing early identification of patients at risk for recurrence. Radionuclide bone scans with technetium-99m (99mTc)-labeled bone-seeking phosphate compounds are particularly useful in detection of metastatic bone lesions.19 Fluorodeoxyglucose positron emission tomography (FDG-PET) is also a useful tool in diagnosis and response assessment.21 Telangiectatic osteosarcoma is characterized microscopically by blood-filled spaces divided by septa containing neoplastic sarcomatous cells. Both radiologically and histologically, it is difficult to differentiate from aneurysmal bone cyst. Telangiectatic osteosarcoma accounts for less than 4% of cases of osteosarcoma. Age and anatomic distributions are similar to those in conventional osteosarcoma. On imaging studies, telangiectatic osteosarcoma manifests as a purely lytic lesion with a permeative destructive growth pattern; it usually disrupts the cortex, but with minimal or no periosteal new bone formation, which often is multilayered, in an onionskin pattern. Overall, the radiographic appearance is not that of a typical osteosarcoma, which usually has a mixture of blastic and lytic areas. Indeed, a purely lytic radiographic appearance is a diagnostic requirement for its diagnosis. This pattern can simulate aneurysmal bone cyst.22,23 Histologically, telangiectatic osteosarcoma is very hemorrhagic, similar to the gross appearance of aneurysmal bone cysts. Microscopically, this tumor consists of cystlike spaces divided by septa, which are composed of highly atypical sarcomatous tissue. Unlike aneurysmal bone cysts, these cystic spaces have no endothelial lining, and the tumor cells are in direct contact with areas of hemorrhage.22 In the past, it was thought that telangiectatic osteosarcoma carried a worse prognosis than for conventional osteosarcoma. With appropriate multimodality therapy, however, the outcome is similar to or better than that for conventional osteosarcoma.15 Low-grade intramedullary osteosarcoma is a very rare variant of osteosarcoma, accounting for less than 1% of cases. Most cases are diagnosed during the third decade of life. Its anatomic distribution is similar to that of conventional osteosarcoma, with predilection for the distal femur and proximal tibia. In contrast with conventional osteosarcoma, symptoms typically develop over many months or even years before the patient comes to medical attention. Imaging studies usually show a variable pattern of lytic foci and dense areas, poorly demarcated. Periosteal new bone formation is minimal. Histologically, it shows a predominantly differentiated fibroblastic and osseous component, similar to what is seen in parosteal osteosarcoma (see “Surface Osteosarcomas” below). The differential diagnosis must distinguish this lesion from fibrous dysplasia. Treatment includes a complete resection of the lesion. Incomplete resection invariably results in local recurrence, and dedifferentiation increases with each recurrence.24 Parosteal osteosarcoma is a low-grade tumor that grows predominantly on the surface of long bones, in an exophytic pattern. Because it is derived from the outer layer of the periosteum, it grows without causing elevation of the periosteum or evidence of periosteal new bone formation. Parosteal osteosarcomas account for 3% of all osteosarcomas. They tend to occur in skeletally mature patients, with diagnosis during the third and fourth decades of life. More than 80% of these tumors are located in the distal portion of the femoral shaft, in its posterior aspect, within the superior popliteal area. Because of their slow growth, parosteal osteosarcomas usually manifest as a painless mass. Imaging shows a tumor growing on the surface of the bone, with a broad base, in a mushroom-like fashion. The mass typically is densely mineralized and has lobulated outlines. Microscopically, these tumors are characterized by the presence of a spindle cell fibroblastic component, with variable osteoid production, low mitotic rate, and no atypical features.25,26 The treatment is surgical, and a complete resection is mandatory. Incomplete resections invariably lead to local recurrences, and the risk of high-grade transformation increases with local recurrence.25 The most important adverse prognostic factor in patients with osteosarcoma is the presence of metastatic disease.17 In addition, primary tumor location is associated with outcome. Children with primary tumors of the tibia and distal femur appear to have a more favorable prognosis than those with axial primary tumors. This finding highlights the importance of complete surgical resection in the management of this malignant disease.17 For patients with localized disease, factors associated with poor prognosis include measures of tumor burden, such as tumor size, and levels of alkaline phosphatase and LDH,17,18,27 as well as more biological measures, such as poor histologic response to preoperative chemotherapy,28 hyperdiploidy,29 and increased expression of P-glycoprotein30 or Ki-67.31 The percentage of tumor necrosis after preoperative chemotherapy is the most consistent and important factor associated with outcome in children and adolescents with localized osteosarcoma. A favorable response (more than 90% tumor necrosis) correlates with excellent overall survival. Patients who have less than 90% tumor necrosis are considered poor responders, for whom the prognosis usually is poor.17,28,32 Because of this strong correlation between degree of histologic response to preoperative chemotherapy and outcome, noninvasive methods such as DEMRI, used for monitoring tumor response, can be used to evaluate changes in regional contrast access during chemotherapy.20 Finally, a proportion of patients with extremity osteosarcoma are seen with a pathological fracture, or a pathological fracture develops after institution of therapy. This has been considered a poor prognostic factor and an indication for immediate amputation. It is possible, however, that with the use of preoperative chemotherapy and judicious use of limb-sparing techniques, a selected group of patients with pathological fracture may still do well without amputation. Optimal management of osteosarcoma consists of multiple-agent chemotherapy and local control measures, including amputation or limb-sparing surgical procedures. Box 95-1 outlines the standard approach to management of osteosarcoma used at most institutions. Before the development of limb-sparing procedures, amputation was the standard surgical method used for curative treatment of osteosarcoma. Amputation now generally is reserved for primary tumors deemed unresectable. Limb function after below-the-knee amputation usually is excellent. Over the past several years, the role of limb-sparing procedures has increased dramatically. As a result of refinements in neoadjuvant chemotherapy, bioengineering, and imaging techniques, it is estimated that as many as 80% of patients with osteosarcoma will eventually be candidates for limb-sparing procedures.33 The criteria for limb-sparing procedures include (a) absence of major neurovascular involvement by tumor, (b) feasibility of wide surgical excision to include a normal muscle cuff in all directions and en bloc removal of all biopsy sites, (c) resection of the adjacent joint and capsule, (d) adequate motor reconstruction with regional muscle transfer, and (e) adequate soft-tissue coverage.34 In the past, immature skeletal age and primary tumor of the humerus were relative contraindications; however, new expandable prosthetic devices may help overcome this problem.33,35 More recent improvements have been concentrated on achieving noninvasive extension of prostheses. One such method is the Phenix technology (Repiphysis). The basic principle involves storage of energy in a spring maintained in compressed form by a locking system. Prosthetic lengthening is performed through exposure to an external electromagnetic field that pilots the locking system and allows controlled release of the spring energy (Fig. 95-3). Prosthetic expansion of several millimeters can be achieved with each procedure, and the total duration of the procedure is less than 30 seconds (Fig. 95-4).36 Thus limb-sparing procedures are considered feasible in the care of most children and adolescents with osteosarcoma. When these procedures are appropriately performed, the risk of local recurrence is low (less than 5%).37 Long-term functional outcome, however, must be carefully compared with that obtained with amputation alone. Complications of limb-sparing surgery include infection, nonunion, fracture, and unstable joints. Before the introduction of adjuvant chemotherapy, fatal metastatic disease developed in more than 80% of patients with osteosarcoma.38 Trials of single-agent chemotherapy began in the 1960s and early 1970s and established, in a nonrandomized manner, a role for the use of chemotherapy in the management of osteosarcoma. Responses with single-agent high-dose methotrexate or doxorubicin occurred in 20% to 40% of patients with metastatic disease.38,39 Since then, different combinations of platinum compounds, doxorubicin, and high-dose methotrexate have formed the basis of standard chemotherapy regimens that lead to cure in 50% to 75% of patients with nonmetastatic disease (Table 95-1). Table 95-1 Summary of Most Relevant Chemotherapy Protocols for the Management of Osteosarcoma *Postoperative regimens are either the same as or in addition to preoperative regimens, or as specified. For more than 2 decades, therapy for nonmetastatic osteosarcoma has followed the basic guidelines of the T-10 protocol,40 and most of the current treatment strategies have evolved from lessons learned with it. The T-10 protocol and its variants consist of a multiple-agent regimen of high-dose methotrexate, doxorubicin, cisplatin, and a combination of bleomycin, cyclophosphamide, and dactinomycin. When the T-10 protocol guidelines are used, the 5-year actuarial event-free survival (EFS) rate may approach 70%.27,28 These results are not always reproducible, however, and multiinstitutional U.S. and European studies conducted according to similar guidelines have shown lower EFS rates, usually 50% to 60%.41–45 The lack of reproducibility of the results of the original study may be attributed in part to the complexity of the treatment. In a study performed by the European Osteosarcoma Intergroup, patients were randomly assigned to receive the T-10 protocol or a much simpler treatment of 6 courses of a combination of cisplatin and doxorubicin.41 Only half of the patients in the T-10 protocol-like group completed all scheduled therapy, whereas 94% of the patients in the short-treatment group received the 6 courses of chemotherapy. The results for both treatment groups were identical, suggesting that a simple, two-drug regimen of cisplatin and doxorubicin can cure more than half of patients with nonmetastatic osteosarcoma.46 A major contribution of the T-10 protocol and its predecessor T-7 is that the histologic response to neoadjuvant chemotherapy has been identified as the most important prognostic factor in the care of patients with nonmetastatic disease.28 Intensification of postoperative28,42,43 or preoperative27 chemotherapy with cisplatin, doxorubicin, and ifosfamide, however, has not improved outcome. To increase the proportion of good histologic responders, some researchers have investigated intraarterial administration of cisplatin. In the context of an aggressive, multiple-agent treatment, however, intraarterial infusion of cisplatin does not offer any significant advantages.47 In recent years, ifosfamide has been incorporated into therapeutic regimens for osteosarcoma. After early reports showed that ifosfamide as a single agent achieved response rates of 10% to 60% in patients with advanced or treatment-refractory osteosarcoma,48,49
Pediatric Solid Tumors
Osteosarcoma
Epidemiology
Tumor Biology
Pathology
Clinical Manifestations
Laboratory and Radiologic Evaluation
Osteosarcoma Subtypes
Telangiectatic Osteosarcoma
Low-Grade Intramedullary Osteosarcoma
Surface Osteosarcomas
Prognostic Factors
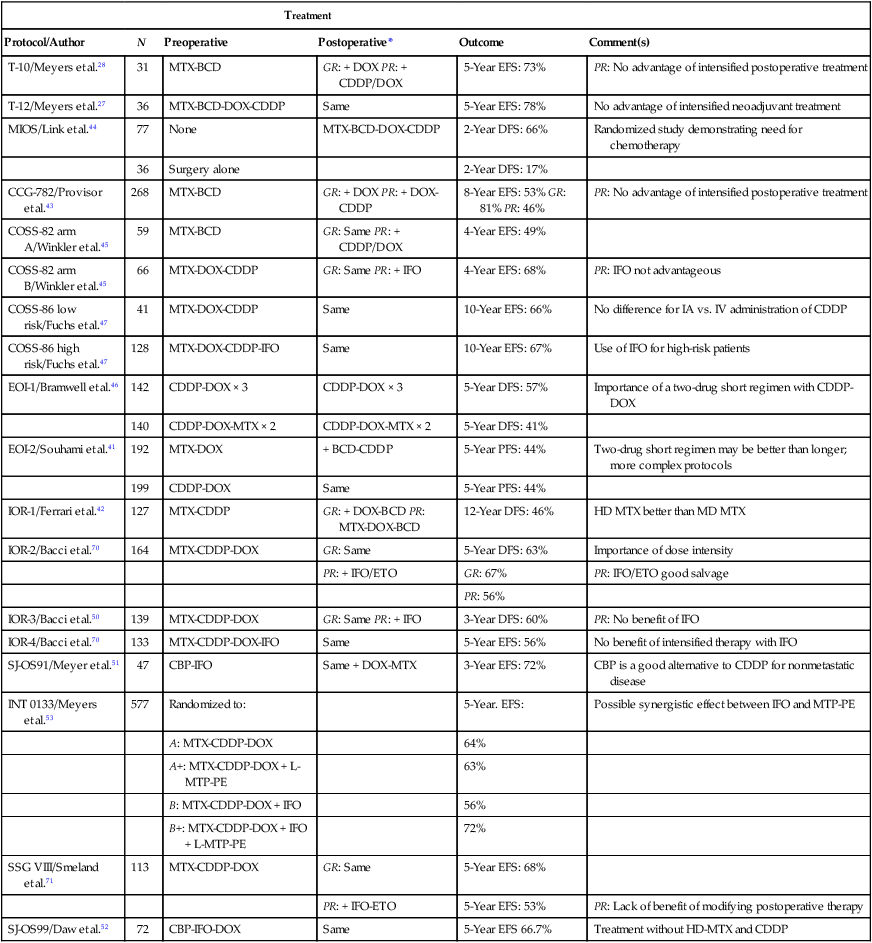
Treatment

Treatment
Protocol/Author
N
Preoperative
Postoperative*
Outcome
Comment(s)
T-10/Meyers et al.28
31
MTX-BCD
GR: + DOX PR: + CDDP/DOX
5-Year EFS: 73%
PR: No advantage of intensified postoperative treatment
T-12/Meyers et al.27
36
MTX-BCD-DOX-CDDP
Same
5-Year EFS: 78%
No advantage of intensified neoadjuvant treatment
MIOS/Link et al.44
77
None
MTX-BCD-DOX-CDDP
2-Year DFS: 66%
Randomized study demonstrating need for chemotherapy
36
Surgery alone
2-Year DFS: 17%
CCG-782/Provisor et al.43
268
MTX-BCD
GR: + DOX PR: + DOX-CDDP
8-Year EFS: 53% GR: 81% PR: 46%
PR: No advantage of intensified postoperative treatment
COSS-82 arm A/Winkler et al.45
59
MTX-BCD
GR: Same PR: + CDDP/DOX
4-Year EFS: 49%
COSS-82 arm B/Winkler et al.45
66
MTX-DOX-CDDP
GR: Same PR: + IFO
4-Year EFS: 68%
PR: IFO not advantageous
COSS-86 low risk/Fuchs et al.47
41
MTX-DOX-CDDP
Same
10-Year EFS: 66%
No difference for IA vs. IV administration of CDDP
COSS-86 high risk/Fuchs et al.47
128
MTX-DOX-CDDP-IFO
Same
10-Year EFS: 67%
Use of IFO for high-risk patients
EOI-1/Bramwell et al.46
142
CDDP-DOX × 3
CDDP-DOX × 3
5-Year DFS: 57%
Importance of a two-drug short regimen with CDDP-DOX
140
CDDP-DOX-MTX × 2
CDDP-DOX-MTX × 2
5-Year DFS: 41%
EOI-2/Souhami et al.41
192
MTX-DOX
+ BCD-CDDP
5-Year PFS: 44%
Two-drug short regimen may be better than longer; more complex protocols
199
CDDP-DOX
Same
5-Year PFS: 44%
IOR-1/Ferrari et al.42
127
MTX-CDDP
GR: + DOX-BCD PR: MTX-DOX-BCD
12-Year DFS: 46%
HD MTX better than MD MTX
IOR-2/Bacci et al.70
164
MTX-CDDP-DOX
GR: Same
5-Year DFS: 63%
Importance of dose intensity
PR: + IFO/ETO
GR: 67%
PR: IFO/ETO good salvage
PR: 56%
IOR-3/Bacci et al.50
139
MTX-CDDP-DOX
GR: Same PR: + IFO
3-Year DFS: 60%
PR: No benefit of IFO
IOR-4/Bacci et al.70
133
MTX-CDDP-DOX-IFO
Same
5-Year EFS: 56%
No benefit of intensified therapy with IFO
SJ-OS91/Meyer et al.51
47
CBP-IFO
Same + DOX-MTX
3-Year EFS: 72%
CBP is a good alternative to CDDP for nonmetastatic disease
INT 0133/Meyers et al.53
577
Randomized to:
5-Year. EFS:
Possible synergistic effect between IFO and MTP-PE
A: MTX-CDDP-DOX
64%
A+: MTX-CDDP-DOX + L-MTP-PE
63%
B: MTX-CDDP-DOX + IFO
56%
B+: MTX-CDDP-DOX + IFO + L-MTP-PE
72%
SSG VIII/Smeland et al.71
113
MTX-CDDP-DOX
GR: Same
5-Year EFS: 68%
PR: + IFO-ETO
5-Year EFS: 53%
PR: Lack of benefit of modifying postoperative therapy
SJ-OS99/Daw et al.52
72
CBP-IFO-DOX
Same
5-Year EFS 66.7%
Treatment without HD-MTX and CDDP
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pediatric Solid Tumors