54 Pediatric Leukemias and Lymphomas
Leukemia is the most common form of malignancy seen in the pediatric age group.1 The vast majority of childhood leukemia cases are acute, unlike those in adults. Acute lymphoblastic leukemia (ALL) is most common, accounting for 80% of all cases of childhood leukemia. Acute nonlymphoblastic leukemia, most often acute myelogenous leukemia (AML), constitutes almost 20% of cases, and chronic myelogenous leukemia (CML) approximately 3% of all leukemia cases.
Over the past two decades, an unexplained increase in leukemia rates among children younger than 15 years of age has been observed in the United States. This trend results primarily from an increase in ALL incidence, since the rates of leukemias other than ALL did not appear to increase from 1977 to 1995.2 Linet et al.3 conclude that there was no consistent change in the incidence of childhood leukemia, but rather an isolated abrupt rise in the mid-1980s. Whether these temporal trends are real events of epidemiologic importance or are merely related to changes in diagnostic and reporting methods is being debated and explored in the next generation of epidemiologic investigations.
Acute Lymphoblastic Leukemia
Epidemiology and Etiology
ALL accounts for more than 2400 of the 3250 new cases of childhood leukemia diagnosed each year in the United States.1 In the United States, childhood ALL has a peak incidence between 3 and 5 years of age, and is more common in boys than in girls and in Whites more than in African Americans.1 The frequency of ALL is increased in certain genetic disorders including Down syndrome, congenital immunodeficiency diseases (e.g., Wiskott-Aldrich syndrome, congenital hypogammaglobulinemia), and chromosomal fragility syndromes (e.g., Fanconi’s anemia, ataxia telangiectasia).4 Molecular studies of identical twins and neonatal Guthrie cards suggest a prenatal origin for acute leukemia, both lymphoid and myeloid subtypes.5,6 A number of environmental factors have been implicated with increased risk, including in utero x-ray exposure, maternal alcohol use and smoking during pregnancy, parental solvent or radiation exposure, postnatal infection and exposure to electromagnetic fields; however, none of these has been identified as a significant single etiologic agent.7
Clinical Presentation
The common presenting signs and symptoms reflect the degree of bone marrow compromise, the extent and location of leukemic cell infiltration, and the general systemic effects of these processes. Fever, the most common feature, is caused most often by the leukemia itself rather than by infection.8,9 Bleeding symptoms with bruising or petechiae and pallor with fatigue are among the common complaints resulting from the thrombocytopenia and anemia, respectively. Bone pain is common due to leukemic periosteal infiltration and marrow expansion. Lymphadenopathy and hepatosplenomegaly result from extramedullary infiltration of leukemic cells, occurring in 60% of children with ALL.8,9 An anterior mediastinal mass is present in up to 10% of newly diagnosed patients, making a chest roentgenogram crucial in the initial evaluations. Central nervous system (CNS) involvement occurs in about 5% of patients at diagnosis. Children younger than age 2 years and those with T-cell ALL have a higher incidence of CNS leukemia. Testicular leukemia manifesting as a painless enlargement of one or both testes is rare, occurring in less than 5% of patients.
Laboratory data may reveal a mild to moderate degree of anemia and thrombocytopenia along with white blood cell (WBC) counts that may be normal, decreased, or increased. A relatively small proportion of patients (14%) have hyperleukocytosis with a WBC count greater than 100,000/mm3.9 Blasts frequently, but not always, are found in the peripheral blood smear.
Diagnosis and Extent of Disease Evaluation
A thorough physical examination is required to determine the extent of the disease and detect lymphadenopathy, abdominal organomegaly, and testicular involvement. CNS involvement is suspected in the presence of cranial nerve palsies, papilledema, a history of headaches, or vomiting, although asymptomatic pleocytosis is the most common presentation of CNS ALL. The traditional definition of CNS disease requires the presence of lymphoblasts in a cytocentrifuged sample of cerebrospinal fluid (CSF) along with at least 5.0 WBC per µL of CSF or the presence of a cranial nerve palsy.10 Studies to evaluate the clinical significance of lymphoblasts in diagnostic CSF samples with less than 5.0 WBC/mL or in the setting of a traumatic tap contaminated with peripheral blood have been inconclusive, and therefore patients are treated as CNS positive.11,12 In addition, the finding of blast cells in CSF with fewer than 5.0 WBC/mL obtained during treatment is not conclusive of CNS relapse.13 Chest roentgenograms are essential to document mediastinal mass and potential risk of airway or vascular compromise before invasive procedures, sedation, or definitive therapy.
Classification
It is now well recognized that pediatric ALL is a heterogeneous disease. Originally, lymphoblasts were classified solely by morphologic criteria and cytochemical staining characteristics. These methods were sufficient to distinguish ALL from AML. More recently, classification of ALL has expanded to include immunologic and cytogenetic criteria. These criteria are useful in defining lineage and maturational stage of the leukemic clone for prognostic and therapeutic purposes as well as contributing to our understanding of important mechanisms of leukemogenesis. Classification of the different types of ALL with the characteristic phenotypes and associated immunologic and cytogenetic features is presented in Table 54-1.
Morphology
In 1976, the French-American-British Cooperative Group (FAB) devised a classification system for ALL based on the morphologic features of bone marrow lymphoblasts.14 This system divided ALL into three subtypes (L1, L2, and L3). Application of this system has been limited, because the distinction between the most common subtypes, L1 and L2 (98% of cases), is somewhat arbitrary, as results are difficult to reproduce between different observers and neither subtype correlates with immunologic, genetic, or clinical features.15,16 This stands in contrast to the L3 subtype, which predicts the mature B-cell ALL phenotype and translocations involving the MYC oncogene on chromosome 8. Thus, excluding the L3 subtype, the FAB classification system for ALL has been replaced by immunophenotyping and cytogenetics-based systems.17
Cytochemistry
Cytochemical staining properties of blast cells can be used to distinguish ALL from AML.18 Myeloperoxidase staining detects myeloperoxidase in the granules of neutrophilic, eosinophilic, and monocytic precursors. Lymphoblasts are uniformly negative, making this an important distinguishing feature of ALL versus AML. Terminal deoxynucleotidyl transferase (TdT), a DNA polymerizing enzyme, is specific to lymphoblasts of precursor B-cell or T-cell origin; mature B-cell lymphoblasts are negative, as are myeloid precursors. The lack of B- or T-cell lineage-specific cytochemical staining characteristics has limited the usefulness of these techniques. In clinical practice, they have been largely replaced by immunologic methods.
Immunophenotype
Normal hematopoietic cells undergo changes in expression of cell differentiation genes recognized as cell surface antigens as they mature from stem cells into cells of a distinct lineage. The pattern of expression of these antigens detected by flow cytometry with a panel of lineage-associated antibodies is called immunophenotype. The immunophenotype of ALL confirms the lymphoid origin and indicates lineage (B- or T-cell), as well as stage of lymphoid maturation at which the malignant transformation occurred. Current therapeutic strategies for ALL require the immunophenotype as part of the diagnostic evaluation.19
Immunophenotype studies of cell surface antigens have defined four subsets of ALL: early pre-B, pre-B, mature B-cell, and T-cell. As detailed in Table 54-1, each immunologic subtype is associated with specific clinical and cytogenetic features. The majority of ALL cases are of immature B-lineage expressing the common ALL antigen, CALLA or CD10. T-cell ALL can also be subclassified according to the stage of thymocyte maturation; however, for therapeutic purposes, only the differentiation between T-cell, mature B-cell, and B-precursor ALL is important for risk stratification and treatment assignment. More recently, measurements of these panels of immunologic markers have been adapted to allow very sensitive monitoring of minimal residual disease.20
Cytogenetics
Improvements in cytogenetic and molecular technologies now make it possible to identify abnormalities in chromosome number or structure in more than 90% of ALL cases.21 The most common abnormality, hyperdiploidy, is associated with B precursor ALL and correlates with a good prognosis. A number of these abnormalities are detectable only at the molecular level; the TEL/AMLI fusion t(12;21) is an example of such an abnormality.22 Cytogenetics and molecular genetics play an important role in the classification of leukemias and correlate significantly with the outcome of the treatment.23 It is now well recognized that the morphology, immunophenotype, and drug responsiveness of the leukemic blast cell are a direct reflection of its genetic characteristics.24,25
Prognostic Factors
Interest in prognostic factors arose in the late 1970s as therapy became successful in many patients who had ALL. Pediatric oncologists began looking for common features among groups of patients who did well compared with patients whose disease relapsed. Through retrospective analysis of disease-free survival (DFS), certain features present at the time of diagnosis of ALL were identified that were useful in predicting which patients had a good, fair, or poor prognosis.10,26,27 The initial WBC count and the age of the child at diagnosis have been the two most reliable indicators of prognosis, both for duration of remission and survival.26,28
Higher leukocyte counts, more frequent in T-cell disease and infants, are strongly correlated with a worse prognosis, although the biologic rationale for this is not clear. Children who are younger than 1 year of age or older than 10 years of age have a relatively poor prognosis compared to those in the intermediate age group. The majority of infants have 11q23 rearrangements, primarily the t(4;11) translocation, which creates an MLL-AF4 fusion gene and is associated with hyperleukocytosis and a poor prognosis.29
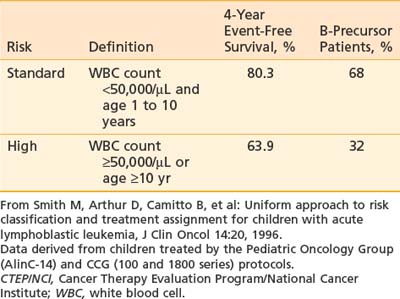
The prognostic importance of other clinical features such as gender, race, degree of organomegaly, presence of a mediastinal mass, initial platelet count, and DNA index is not reproducible among different study groups. As a result, the National Cancer Institute (NCI) sponsored a workshop in September 1993 to define uniform criteria to be used for risk-based treatment assignment for children with ALL. Workshop participants agreed on uniform age and WBC count criteria (Table 54-2) and a common set of prognostic factors to be evaluated prospectively including cytogenetics, early response to treatment, and minimal residual disease status.28 Current ALL risk classification schemas of the major cooperative groups use these NCI criteria for age and WBC count to define standard- and high-risk groups, and for purposes of treatment intensity many groups further subdivide into a low-risk group if they have favorable cytogenetics such as hyperploidy, trisomies 4, 10, and 17 or telAML1, and early response to therapy (such as a day 15 marrow) and low minimal residual disease (MRD) measured at the end of induction. Patients are considered higher risk if they have CNS or testicular disease at diagnosis. A very high-risk group is defined by the presence of MLL gene rearrangement, Philadelphia chromosome, minimal residual disease at the end of induction, or induction failure.30
Risk-Targeted Therapy
The Childhood Cancer Survivor Study recently published the results of their 25-year follow-up among 4,000 survivors of ALL diagnosed before the age of 21 years.31 Significant findings include an excess cumulative mortality of 13% due to recurrent ALL and second neoplasms. In comparison to the general population and a sibling cohort, survivors reported more frequent chronic health problems, mental health issues, and functional limitations with lower rates of college graduation, employment, and marriage. The risk of death or adverse outcome was greatest among survivors treated with radiation. These results highlight the need for ongoing multidisciplinary care for the growing numbers of leukemia survivors, but also the need for continued research and refinements of therapy to avoid such late effects.
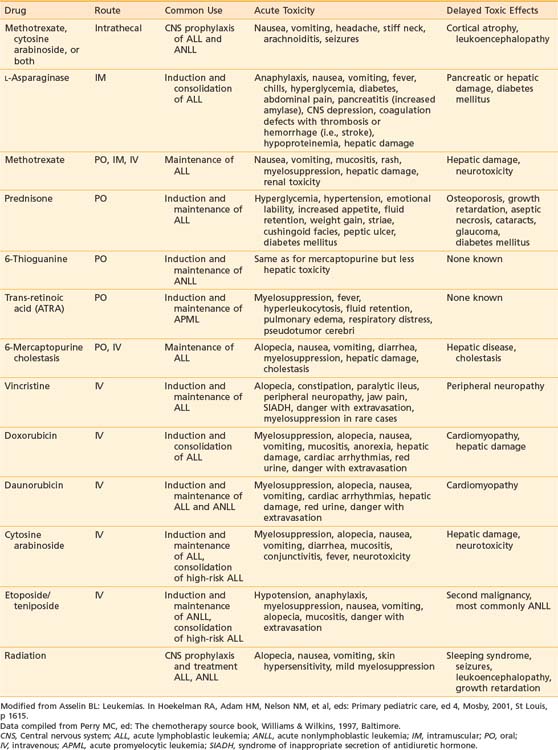
Common Elements of Therapy
Most treatment regimens for B-precursor ALL divide therapy into four phases: initial remission induction, intensification (consolidation), CNS prophylaxis, and continuation (maintenance) therapy. Induction treatment typically includes prednisone, vincristine, and asparaginase and intrathecal therapy. With these drug regimens, complete remission is achieved in 97% of children.32 Treatment for prevention of overt CNS disease is initiated early and continues at intervals throughout the other phases of therapy. Most centers continue treatment for 2.5 to 3 years, which includes 1 to 2 years of maintenance therapy with nercapto, mtx, and variable number of pulses of vincristine and corticosteroids.
Mature B-Cell ALL
Similar to Burkitt’s lymphoma, mature B-cell ALL is more common in boys, with a median age of onset of 8 years. It frequently is associated with bulky abdominal tumor and a very high rate of cell proliferation, factors that can result in the metabolic complications associated with tumor lysis syndrome. When treated with traditional ALL therapy, patients with B-cell ALL had a grim prognosis. This changed dramatically with the development of short-term (6- to 8-month) regimens of extremely intensive therapy that incorporate fractionated high-dose cyclophosphamide, high-dose methotrexate, and cytarabine.33–35 These regimens have been adopted by virtually all centers with event-free survival (EFS) of up to 80%. Some groups are looking at the benefits of addition of ifosfamide, etoposide, or both in these regimens36,37 but at the cost of significant toxicity. Rituximab, a chimeric monoclonal antibody to CD-20, has shown remarkable response rates when added to chemotherapy of adults with non-hodgkins lymphoma (NHL) without additional toxicities. The efficacy, toxicity, and pharmacology are currently being evaluated in children with B-cell disease in a Children’s Oncology Group (COG) trial.
T-Cell ALL
Historically, T-cell ALL in children has been associated with a worse prognosis than in other subtypes of childhood ALL. Patients with T-ALL are more likely to present with high-risk features of older age, high leukocyte count, mediastinal mass, bulky organomegaly, and lymphadenopathy often termed “lymphomatous presentation,” as well as testicular involvement. T-cell ALL can be successfully treated according to the same regimens as B-lineage ALL, although, regardless of presence of other risk features, these patients require treatment on a high-risk arm.38,39 High doses of methotrexate (5 grams/m2) appear to improve the outcome in patients with T-cell ALL.38,40 Nelarabine, a prodrug of Ara G, has been shown in early phase clinical trials to have selective cytotoxicity for T-lineage lymphoblasts and has been incorporated into an ongoing study of COG to evaluate its safety and efficacy when combined with multiagent chemotherapy.41
ALL in Infants
Among children younger than 1 year, in whom the prognosis is poor, 80% have rearrangements of the MLL gene.27,42 Most cooperative groups have designed treatment regimens specifically for infants, which include high-dose cytarabine, high-dose methotrexate, etoposide, and cyclophosphamide combined with elements of high-risk ALL therapy such as vincristine, corticosteroid, asparaginase, and anthracyclines. Allogeneic bone marrow transplantation (BMT) is considered in first remission when a matched-related donor is available. Overall, the cure rates are low with DFS of 40% to 60%. Recent biologic studies of cases of infant ALL with MLL gene rearrangements have shown high levels of FLT 3 expression. This observation has created great interest in development of FLT 3 tyrosine kinase inhibitors such as lestaurtinib (CEP 701) as a treatment to improve outcome in infant ALL.43,44
The Evolving Role of Radiotherapy
Historically, the role of radiotherapy in ALL has been in the management of extramedullary disease. Trials that compared its efficacy to that of alternative therapy for both prophylaxis and treatment of extramedullary leukemia addressed the growing concern about the potential long-term toxic effects of radiation in young children.45–48 The findings of these trials have resulted in a reduction in the use of radiotherapy in the treatment of extramedullary pediatric ALL. More recently, radiation has played a significant role in BMT cytoreductive therapy designed for patients with a poor prognosis or relapse.
Central Nervous System Prophylaxis
With the advent of effective CNS prophylaxis, the survival rates associated with ALL improved dramatically. Improving the quality of life in the increasing number of survivors became a main focus of CNS prophylaxis trials of the 1980s. Ways to modify the delivery of CNS radiotherapy to potentially decrease the incidence and severity of treatment-related late toxicity were explored. Reduction of the cranial radiation therapy (CRT) dose from 24 Gy to 18 Gy was tested successfully in the Children’s Cancer Group (CCG) 143 trial (1974 to 1975).49
Of note is a recent collaborative meta-analysis of pre-1993 trials performed to clarify the relative effects on relapse and survival of different types of therapies directed at the CNS in childhood ALL.50 Radiotherapy and long-term intrathecal therapy gave similar outcomes, with no significant difference in EFS despite random assignment of treatment to 2848 patients, 1001 of whom suffered relapse or death. Intravenous methotrexate reduced non-CNS rather than CNS relapses, and hence the addition of intravenous methotrexate to a treatment regimen including radiotherapy or long-term intrathecal therapy improved EFS with a 17% reduction in the event rate. The EFS at 10 years in these trials was 61.9% without intravenous methotrexate and 68.1% with intravenous methotrexate. There was no significant difference in survival. No evidence was found regarding differences, between trials or between subgroups of different types of patients, in the relative effects of treatment. The conclusion was that radiotherapy could be replaced by long-term intrathecal therapy, and intravenous methotrexate gives some additional benefit by reducing non-CNS relapses. Both of these strategies have become common practice so that CRT is reserved for CNS prophylaxis in limited numbers of cases considered at high risk of CNS relapse because of young age, T-cell disease, high WBC count, or lymphomatous presentation. Additional strategies to further reduce the potential for late morbidity associated with CRT include dose reduction to 12 Gy51 and hyperfraction,52,53 but current investigative efforts have centered principally on finding effective strategies for CNS prophylaxis that do not require the use of radiotherapy.54–61
In current practice, CRT has been eliminated from CNS prophylactic therapy regimens in patients with B-precursor ALL who are between the ages of 1 and 9 years, with an initial leukocyte count of less than 50,000/mL and no evidence of lymphomatous or CNS involvement. The elimination of CRT in the treatment of “higher-risk” patients (aged >9 years and/or WBC count >50,000) remains a controversial issue. In this setting, the need for CRT as part of prophylaxis is highly dependent on the intensity of intrathecal medications and systemic medications that have CNS activity62–65 and which are administered as part of the chemotherapy regimen.
Meningeal Leukemia
Craniospinal axis radiotherapy has been shown to be successful in controlling meningeal leukemia. However, its associated toxicities both to the CNS and to the hematopoietic tissue (producing immunosuppression and thus a compromise in systemic chemotherapy delivery) have prompted investigators to develop other treatment strategies aimed at decreasing the role of radiotherapy, either by eliminating it entirely or by modifying the dosing regimen or volume, or both. Attempts at curing meningeal leukemia with chemotherapy alone, initiated in the 1970s, have generally been unsuccessful.66 Historically, regardless of the drug combination or route of administration employed, there was no convincing evidence to support the use of chemotherapy alone in the eradication of overt CNS leukemia. Although excellent remission rates are achieved with chemotherapy (both intrathecal and systemic), they were usually of short duration.66–68 However, recent data suggest that children diagnosed with B-ALL and CNS involvement (defined as L3 CSF blast, cranial nerve palsy, clinical spinal cord compression, isolated intracerebral mass, or cranial or spinal parameningeal extension) can be cured with standard intensity French-American-British/Lymphoma Malignancy B (FAB/LMB) therapy with the addition of high-dose methotrexate (8 g/m2) plus extra intrathecal therapy.69 Yet, patients with CNS involvement at diagnosis did have an inferior event-free and overall survival (OS) compared to those without CNS disease. Confirmatory trials are in progress.
On the basis of the results obtained to date for chemotherapy alone, CNS radiotherapy should be considered in any regimen designed for the treatment of overt meningeal leukemia, regardless of previous CNS radiation exposure, with the caveat that certain chemotherapeutic approaches may obviate the necessity of RT.35 Since the number of leukemic patients who exhibit meningeal disease is small, and since the systemic and intrathecal chemotherapy employed in the trials designed to deliver CNS radiotherapy is variable, definitive conclusions cannot yet be made with regard to the optimal dosing regimen (hyperfractionated70 versus conventional radiotherapy), field extent (craniospinal versus CRT), and timing (early versus delayed) of the radiotherapy for all scenarios of CNS disease presentation.
Recent reports of success in patients who present with CNS disease and are treated without any RT,69 with low-dose CRT (6 Gy)71 or no spinal radiation at all,72 however, do clearly challenge the necessity of spinal irradiation in meningeal leukemia, especially for chemotherapy-naive patients in whom drug-resistant clones have not yet developed. For this group of patients, CRT of 18 to 24 Gy has become a widely accepted form of CNS radiotherapy, although controversy still exists about the appropriate timing of the delivery of CRT.11 For those patients who develop a relapse either during or off therapy, CRT (craniospinal axis radiation) remains the most widely used form of CNS radiotherapy.73,74 The commonly employed total dose for the CRT is 18 to 24 Gy. Although some investigators suggest that a spinal dose of 18 Gy may be necessary for optimal CNS control, there is sufficient evidence to support the use of a lower dose of 12 Gy.75,76
A recently reported Pediatric Oncology Group (POG) trial successfully treated patients in first remission with isolated but delayed (CR1 ≥18 months) CNS relapses of B-ALL with intensive systemic chemotherapy and delayed RT (18 Gy cranial), whereas patients who relapsed within 18 months received CSI (24 Gy cranial and 15 Gy spinal).77 Currently, there is an ongoing COG study in patients with late (>18 months from diagnosis) isolated CNS relapse to evaluate the effects of intensified systemic chemotherapy with delayed reduced dose cranial radiation of 12 Gy given at 1 year postrelapse. Almost all of these patients will have been treated with systemic and intrathecal (IT) chemotherapy as primary CNS preventive therapy since RT is reserved for ∼10% to 15% of patients with highest-risk disease.78
For the small group of patients who present with symptomatic cranial nerve deficits, an immediate course of CNS radiation is a consideration. Since the goal of such therapy is to salvage nerve function, the use of as low a dose as possible to produce the desired effect is advocated. Although irradiation of the entire cranial contents is often used for this purpose, treatment to a more limited field, to include only the base of the skull, to total doses of 10 to 15 Gy, is recommended. If CRT is used, it is important that the patient be treated in a prone position, if subsequent craniospinal irradiation treatment is to be administered in that position. However, newer RT techniques, pioneered in the setting of brain tumors such as medulloblastoma, include IMRT and tomotherapy, which involve patients being treated in the supine position.79,80,80a Completion of the curative CNS radiotherapy, that is, craniospinal irradiation, may be initiated once hematologic remission has been achieved and the chance of reseeding of the CNS from medullary leukemia has been reduced. After low-dose emergent radiotherapy, an additional course of 12 to 18 Gy to the cranial contents is administered.
Techniques of Central Nervous System Radiotherapy
Cranial Radiation Therapy
When CRT is indicated, the intent is to deliver a full dose to the subarachnoid spaces, including the superficial meninges and the majority of the retina and orbital apex. By providing a 1- to 2-cm field margin around the skull and restricting the beam energy to 6 MeV or less (necessary for treatment of the lateral meninges, which are close to the skull surface), one can deliver a full dose as planned with opposed lateral photon beams.81 To assure adequate margin inferiorly on the base of the skull, the bottom edge of the second cervical vertebra is used. Special attention is needed when designing the blocking to ensure an adequate margin on the cribriform plate and temporal fossa. Because the subarachnoid space extends around the optic nerves and the retina is anatomically an extension of the nervous system, both can be involved by leukemia independently or in association with other CNS sites. Consequently it is standard practice to include the optic nerves, posterior retina, and orbital apex in the treatment field. Several techniques allow one to encompass the posterior orbit and globe while sparing the sensitive anterior aspect of the globe and lens. One approach uses inferior rotation of the gantry (i.e., angling the beam posteriorly) to achieve a parallel anterior margin at the bony orbital rim. The block or multileaf collimator margin for this approach is identified by fiducial field markers placed at the bony orbital rim during simulation, or simply by identifying this structure during a CT simulation.80b
Craniospinal Axis Irradiation
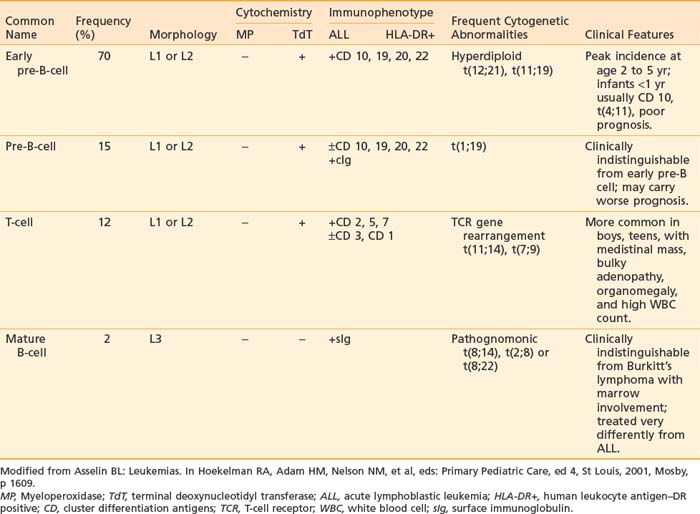
Photon therapy is most widely used for the spine fields; however, electrons may be used when the depth of the anterior surface of the spinal cord is situated within the maximum range of available electrons, after the bone attenuation is accounted for. Although the dosimetry of electrons in regions of inhomogeneities is quite complex, the sparing of anterior structures can be substantial, as shown in a comparison of the dose distributions generated with photon and with electron spinal fields (Fig. 54-1). The spine, indicated on the illustration by a dashed line, is adequately covered using either modality. The electrons, however, deliver less than 10% of the dose anteriorly, whereas the photon beam delivers 70% to 90% of the prescription dose. Depending on the location of the cranium-spine field match planes, the reduction in dose to the thyroid may be substantial. New techniques with the patient supine, and using IMRT, are appropriately employed by experienced teams.79–80 Proton beam therapy for craniospinal irradiation provides even greater sparing of the structures anterior to the spine, at the same time reducing the integral dose of radiation, the importance of which is magnified in children because of late carcinogenic effects of radiation.80 Proton beam therapy is used in the treatment of patients with medulloblastoma and other brain tumors, and is technically feasible for the treatment of the spinal axis in the setting of leukemia.80a
Craniospinal Field Matching
The matching of the posterior spine field with the cranial fields adds technical complexities to the alignment of the cranial fields as discussed for CRT. One approach to handling this junction problem is to accept a slight overlap because of beam divergence (Fig. 54-2A); another is to attempt to create a match plane, accomplished by pedestal rotation, which abuts to both fields (Fig. 54-2B); yet another approach is to create a gap between the cranial and spinal fields (Fig. 54-2C). In all approaches, however, the collimator is rotated for the cranial fields to create a field edge, which follows the divergence of the upper spine field (Fig. 54-2D), and the juncture where all three fields come together is shifted or moved daily to allow for an averaging out of the high-dose and low-dose regions in the junction (feathering). For any cranial field, the inferior border should never be higher than the bottom of C2 to assure complete coverage of the base of the skull; however, caudally it is limited by the presence of the shoulders. In general, the cranial field length should change by 2 to 4 cm each day of a 4-day cycle, which is equivalent to shifting the field edge by 1 to 2 cm daily.
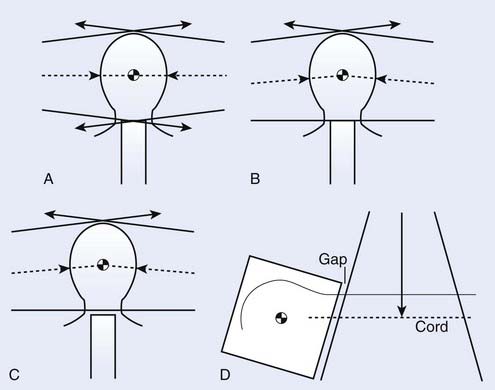
Although gapping is a common practice, it has been shown that improved dose distributions over the junction region are achieved when fields are matched on the skin such that all three fields intersect along the mid-sagittal plane.82 Regardless of whether gapping is used, significant problems with dose inhomogeneity arise from variability in the daily treatment setup.83 When gapping is used, feathering the fields reduces the extent of inhomogeneity resultant not only from errors in the daily setup but also from the inherent underdosage in the gapped region. As noted above, IMRT (including tomotherapy) may obviate the need for such techniques by treating the entire CSI in one field, thereby avoiding the cumbersome problem of matching the fields.
Recommended Technique for Photon Craniospinal Irradiation
The problem of determining a collimator, gantry, and pedestal angle when planning photon craniospinal irradiation is quite complex.84 A recommended approach is to situate the central axes of the lateral cranial fields along the line bisecting the globes (in order to avoid divergence of the field through the contralateral lens); to rotate the collimator of the cranial fields to follow the divergence of the upper spine field (abutting the upper spine field with the cranial fields on the posterior surface of the neck using the light field); and to feather the junction daily.
Electron Spinal Fields
As previously discussed, electrons may be used to treat the spine where the maximum depth of the spine is within the range of the 80% to 90% depth dose of the most energetic electron beam available, including corrections for the bone heterogeneity. The dosimetry of the electron beams must be carefully examined, as algorithms for handling the presence of the bone heterogeneity within the field may use one-, two-, or three-dimensional corrections. With the use of computed tomography scanning to generate isodose curves, it has been reported that the 90% isodose curves shift toward the patient’s posterior surface on the order of 4 mm because of bone absorption and scatter.85 An additional 3-mm geometric margin is added to give a total shift of the 90% isodose line of 7 mm posteriorly.
Testicular Leukemia
Clinically evident testicular disease at diagnosis is a rare event.86,87 Before the advent of modern-day chemotherapy regimens, testicular relapse was a significant obstacle to cure.87,88 Because of the high rate of relapse and because of the high frequency of leukemic infiltration of testes (64% to 92%) demonstrated in autopsy series,89–91 testicular biopsies at completion of therapy became a routine practice for many investigators, as did the delivery of testicular irradiation to those boys with positive findings. Although the approach of administering presymptomatic testicular irradiation to high-risk males reduced the incidence of testicular relapse, the ultimate survival was not significantly improved.48,92–95 Since the introduction of aggressive regimens, not only have DFS rates improved, but isolated testicular relapse has become a rare event.95 As a result, testicular biopsies at completion of therapy in high-risk patients are no longer performed, and prophylactic testicular radiation is no longer necessary.
Confirmation of clinically suspected testicular leukemia is done by wedge biopsy. Bilateral biopsies should be performed, since studies have shown that involvement is bilateral.92,94 At diagnosis, testicular involvement is most often treated with systemic chemotherapy and rarely requires testicular radiation.96 Testicular relapse requires a course of testicular irradiation in combination with intensive salvage systemic chemotherapy. Because of the bilateral nature of testicular leukemia, the target volume should include the entire scrotum to encompass both the testes and epididymis. Although the exact total dose required for local control has not been established, it is clear that doses of less than 12 Gy are suboptimal,88,97 and those of 18 to 24 Gy (at 1.8 to 2 Gy/fraction) are effective.97–100 Reports of prolonged DFS after testicular relapse with aggressive salvage therapy have been encouraging, especially for those patients with an isolated testicular relapse occurring 6 months or longer after completion of therapy.101,102
Testicular radiation at doses used for ALL, independent of any contribution of chemotherapy effect, is associated with sterility and altered Leydig cell function; the latter could result in delayed sexual maturation and the need for appropriate androgen replacement.103,104
Technique of Testicular Radiotherapy
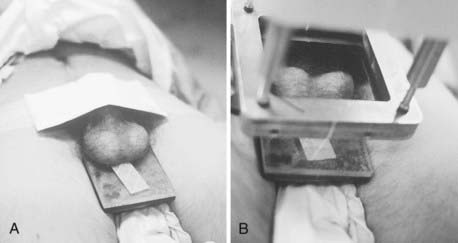
The electron beam, as administered in the energy range most often appropriate for testicular irradiation (9 to 12 MeV), offers the distinct advantage over photon modalities of a region of high-dose uniformity followed by a rapid dose falloff. There should be no more than a 10% variance in dose homogeneity within the treated volume, and the underlying perineal tissues should be spared. A polystyrene/lead combination block is used to support the testicles posteriorly and to shield the perineum (Fig. 54-3A). For electron energies of 9 to 12 MeV, 5-mm lead is adequate for shielding and the overlying 5-mm polystyrene dissipates the effect of electron backscatter. Skin apposition of the beam is achieved by angling the gantry so that the end of the cone is parallel to the treatment surface (Fig. 54-3B).
The Current Era
Recently reported results of major cooperative group studies demonstrate that childhood ALL is currently curable in approximately 80% of cases (Table 54-2). This level of success has resulted from the collaborative efforts of clinical oncologists and laboratory researchers over several decades. Each group has made significant contributions to the design of the sequential clinical trials that have identified increasingly effective treatments and deepened our understanding of the heterogeneous nature of leukemia. The treatment strategies responsible for these high cure rates for ALL are listed in Table 54-3. These successful treatment strategies form the basis for current ALL trials.
Vincristine, prednisone, methotrexate, and mercaptopurine have been the backbone to virtually all successful ALL treatments for 30 years. The dosage and schedule varies among specific protocols. Additional effective antileukemic drugs have been discovered and incorporated into the current regimens. The role and common complications of the common agents used in ALL therapy are listed in Table 54-4. The development of these intensive systemic combination chemotherapy regimens has resulted in marked improvement in outcomes especially in patients presenting with unfavorable immunophenotypic and biologic features.
The New Era: Future Directions
The evolution of curative therapy in ALL continues, with the ultimate goal of increasing the cure rate of all children with ALL, regardless of risk group, while at the same time decreasing the potential for adverse treatment-related sequelae. One direction being taken is to develop more sensitive techniques to detect MRD prospectively in patients who are apparently in complete remission according to current criteria, and thus may benefit from additional therapy to prevent relapse. Another direction is to discover new, effective antileukemic drugs and treatment approaches. Several new anticancer agents are currently being studied for ALL. They include such families as the cytokines (interferon-a, interleukin-4, and tumor necrosis factor) and the antibody-toxin conjugates.120,121 Innovative treatment regimens are being designed to better address the needs of the continuously evolving biologic subtypes and risk groups of ALL, including the use of hematopoietic growth factors to permit further intensification of therapy. Still another direction is to devise effective salvage therapy for those children who do not achieve remission or who relapse on current protocols.
Acute Nonlymphocytic Leukemia
Epidemiology and Etiology
Acute nonlymphocytic leukemia (ANLL) represents 20% of acute leukemias in children, accounting for approximately 650 of the 3250 new cases of childhood leukemia diagnosed each year in the United States.1 This is a striking contrast to the prevalence of ANLL or AML in adults, where it accounts for 80% of acute leukemias or approximately 10,000 new cases per year. The incidence of ANLL remains steady from birth to adulthood with only a slight peak in late adolescence. The median age of onset of ANLL is generally older than that of ALL, approximately 8 to 10 years.
Clinical Presentation and Diagnosis
The clinical presentation of ANLL is very similar to that of ALL. Pallor, fatigue, skin or mucosal bleeding, and fever are present in the majority of children at initial presentation.122 Infection at diagnosis is also common. Bone pain, lymphadenopathy, and marked hepatosplenomegaly are less common in ANLL than in ALL. Chloromas are solid tumor collections of immature myeloid cells that can occur in patients with ANLL. Leukemia cutis is more common in infants than older children. Almost all patients have some degree of anemia and thrombocytopenia. Twenty percent of patients present with hyperleukocytosis with a WBC count greater than 100,000/mm3. Bleeding secondary to a coagulopathy is more common in ANLL and results in an increased risk of early death from hemorrhage, especially intracranial bleeding. This is particularly true of patients with acute promyelocytic leukemia, the majority of whom have a coagulopathy at diagnosis.
The diagnosis of ANLL is confirmed by bone marrow aspirate showing greater than 25% blasts. Cytochemical staining and immunophenotyping of the leukemic blast cells are important to distinguish between the two forms of acute leukemia and help to distinguish among the different subtypes of ANLL (see Table 54-1). In young infants with Down syndrome, true ANLL must be differentiated from transient myeloproliferative syndrome.123 No single test is available to distinguish infants who have this transient myeloproliferative disorder from those who have true congenital leukemia, so conservative therapy with close monitoring for 6 to 8 weeks is the recommended approach in this situation.
Classification
The blast cell population in patients with ANLL can be characterized by morphology, cytochemistry, immunophenotype, and cytogenetics as detailed in Table 54-5. The current system of classification, defined by the FAB working group in 1976, recognizes eight subtypes of ANLL based on degree of cellular expression of granulocytic, monocytic, erythroid, or megakaryocytic features.124
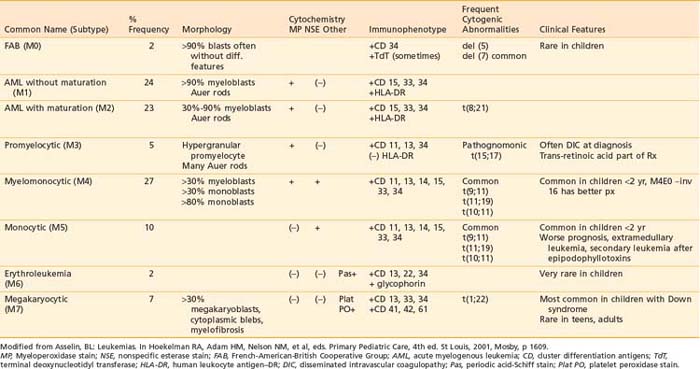
Acute myeloid leukemia (M1 and M2) is the most common form of ANLL, representing almost 50% of all cases of pediatric ANLL.125 Among patients younger than 2 years of age, the majority have either myelomonocytic or monocytic leukemia (M4 or M5 subtype) and frequently present with high WBC counts, leukemia cutis, and CNS disease.126 Acute megakaryocytic leukemia (M7) is seen most frequently in infants, especially those with DS.127
Cytogenetic abnormalities have been observed in the majority of patients with ANLL.128 Of these, the t(15;17) pattern is the only one associated with a particular FAB subtype, namely acute promyelocytic leukemia (M3). The presence of the PML-RARa fusion gene is pathognomic for M3 subtype and is thought to be crucial to pathogenesis as well.129 A unique translocation, t(1;22), has been described in infants with M7 megakaryocytic leukemia.130 The t(8;21), which produces a fusion gene of the AML1 and ETO genes from chromosomes 21 and 8, respectively, is the most common gene abnormality in AML, usually the M2 subtype, and is associated with better remission rates and prognosis.131 Translocations involving the 11q23 band with a breakpoint involving the MLL gene are common in M4 and M5 subtypes, as well as infant leukemias and secondary leukemias following epipodophyllotoxin therapy.132 Deletions of single chromosomes, either 5 or 7, are not specific to an FAB subtype but predict a poor prognosis.128,131
Prognostic Factors
The evolution of curative therapy for ANLL has not progressed as rapidly as that for ALL. Because of the relative scarcity of effective treatment options for patients with ANLL, there has been limited clinical application of the information obtained from immunophenotype or cytogenetics studies in terms of identifying prognostic indicators. Initial leukocyte count, age at diagnosis, and FAB classification appear to be the most significant factors related to treatment outcome. High WBC, age older than 14 years, infancy (age younger than 1 year), and FAB M5 subtype have been associated with worse prognosis in most studies.125,133 The era of tailored therapy to meet the specific needs of different risk groups has not been reached in AML.
Treatment
Chemotherapy
The majority of patients with ANLL are treated according to one standard therapeutic regimen. In contrast to the marked improvements in survival outcome in pediatric ALL, long-term EFS rates in ANLL have not increased dramatically over the past few decades. Although response rates may reach levels of 75% to 85%, the median duration of remission rarely exceeds 2 years.134 Relapse occurs frequently during the first 6 to 12 months on therapy, and early deaths due to treatment-related complications such as sepsis and bleeding reach levels as high as 5% to 15%. The 2-year EFS rate with current therapy remains modest, with reported values of 35% to 45%.135,136 The CCG-2961 trial results were recently published, which demonstrated a 5-year EFS of 42% and OS of 52%. This was a significant improvement in survival compared to the 44% reported between 2000 and 2002, which they attribute to increased experience and decreased toxic deaths, rather than new drugs or treatment strategies.137
Chemotherapy for all subtypes (excluding M3) consists of one to two cycles of induction therapy to achieve complete remission and postremission consolidation therapy for three to six cycles. Maintenance therapy is rarely used as it has shown clear benefit only in acute promyelocytic leukemia. Induction regimens include combinations of cytarabine and an anthracycline such as daunorubicin, mitoxantrone or idarubicin often with etoposide, thioguanine or dexamethasone. The intensity of induction therapy appears to be an important feature in the overall outcome of therapy. These regimens all produce severe myelosuppression, which is considered a key element to induce a complete remission. Current remission rates are 75% to 90%.138–140 Approximately one half of the induction failures are attributed to resistant disease and the other half to toxic deaths. Because of the intensity of therapy used to treat AML, patients should have their care coordinated by specialists in pediatric oncology and should be treated in hospitals with necessary supportive care facilities.
Postremission therapy may utilize either continued intensive chemotherapy with cytarabine-based regimens or allogeneic stem cell transplantation for the subset of patients with a matched related donor. The role of hematopoietic stem cell transplantation (HSCT) in the treatment of AML continues to evolve. Autologous transplantation achieves disease free survival rates of 55% to 60% similar to those observed with chemotherapy alone.141–144 Allogeneic HSCT has demonstrated better outcomes when compared with chemotherapy and autologous transplant, but is limited in its application because the minority of patients have a human leukocyte antigen (HLA)-matched family member to serve as donor. Several recent studies have identified favorable prognostic features that select patients with predicted DFS of 70% to 80% with chemotherapy alone.145–148 In these patients, HSCT would be considered only after a first relapse. Ongoing trials are investigating alternate donor HSCT (unrelated match, unrelated cord blood and haploidentical) for children who have poor-prognosis AML and who lack an HLA-identical sibling donor.
With growing recognition of the importance of morphology, cytogenetics, and immunology in distinguishing subtypes of ANLL with respect to the selection of chemotherapeutic agents, investigators have initiated subtype-specific therapeutic trials. The results of such trials are promising.149–159 At present, allogeneic transplantation is the recommended choice for postremission induction therapy in newly diagnosed patients with ANLL who have an available matched, related donor (unfortunately the minority of patients). The role of allogeneic transplantation using unrelated donor sources is controversial for patients in first remission.
Acute Promyelocytic Leukemia
Acute promyelocytic leukemia (APML) is the only subtype of ANLL for which there is type-specific therapy. The t(15;17) translocation, which characterizes APML blasts, produces a PML/RARa fusion gene resulting in disruption of the retinoic acid receptor and blockade of normal cellular differentiation pathways. All-trans-retinoic acid (ATRA) binds to the chimeric RARa and overcomes the differentiation blockade, resulting in cell maturation, then apoptosis.160,161 The best results have been obtained with the combination of ATRA and chemotherapy with induction rates of 90% and DFS of greater than 70%.161,162 Arsenic trioxide, also a differentiating agent, has been effective in treatment of APML, especially when resistance to ATRA develops.163 Ongoing clinical trials are designed to determine how the differentiating agents, ATRA and arsenic, can best be combined with cytotoxic drugs for the safe and effective treatment of APML.
Down Syndrome and Acute Nonlymphocytic Leukemia
Although rare in children without Down syndrome, megakaryocytic (M7) leukemia is the most common form of AML in young children with the disorder.164 Interestingly, children with Down syndrome and AML have been observed to have better responses and superior outcomes (EFS rate ∼80%) with standard AML therapy than other children with AML, even the M7 subtype. This has been shown to be a characteristic of the patient, not of the leukemia. Most treatment failures in patients with Down syndrome are a result of treatment-related mortality.165,166 Thus, increased intensity regimens and allogeneic BMT are not recommended for these patients.
Secondary Leukemia
The association between treatment with the epipodophyllotoxins, etoposide or teniposide, and development of secondary ANLL is now well established.167 The epipodophyllotoxin-induced leukemias are characterized by a short latency period (2 to 4 years) and a myelomonocytic or monocytic phenotype with a translocation involving band 11q23 with MLL gene rearrangement.168 In contrast, secondary leukemia that develops after therapy with alkylating agents is characterized by a longer latency period (∼10 years), a prodrome of myelodysplasia and monosomy 5 or 7 genotype.169 Secondary leukemias are not responsive to traditional chemotherapy and BMT holds the only hope for cure.
The Role of Radiotherapy
Chloromas, also known as myeloblastomas and granulocytic sarcomas, are extramedullary masses of malignant myeloid cells associated most commonly with ANLL (especially FAB M2), but also observed in patients with CML.170 Doses of 24 Gy, at 2 Gy per fraction, are highly effective in eradicating the local masses of malignant infiltrates refractory to chemotherapy.
Future Therapies
One of the exciting new areas of research is in the use of targeted immunotherapies.121 Gemtuzamab ozogamicin (myeloTang), a humanized anti-CD33 conjugated to calicheamicin, is an example of such an agent. Clinical trials with this immunoconjugate have shown an approximate 37% remission rate in adults with relapsed AML.171,172 In addition, a CD-45 monoclonal antibody has been radiolabeled with iodine-131 for use during BMT as a means of delivering focused bone marrow irradiation.173 Mutation of the Ras oncogene is common in AML. The Ras product must be farnesylated for it to function properly as a growth signal transduction protein. A new class of compounds called farnesyl transferase inhibitors has been developed and clinical trials are under way.174
Chronic Myelogenous Leukemia
CML is rare in childhood, representing 3% to 5% of all pediatric leukemias.1 In childhood, the disorder may appear as two distinct clinical syndromes: adult-type CML, which is virtually indistinguishable from that seen in older patients, and juvenile myelomonocytic leukemia (JMML; formerly known as juvenile CML), a disease relatively restricted to very young children that has distinct clinical, laboratory, and cytogenetic features.175
The classic adult type is characterized by the presence of the Philadelphia chromosome, a reciprocal translocation t(9;22) (q34;q11). The resultant fusion gene formed, BCR/ABL, appears to have a major role in the pathogenesis of CML. It is typically seen in adolescents, with a peak incidence between 10 and 14 years of age. Treatment and outcomes of CML in childhood parallels that of the adult patient.176 Allogeneic BMT is the only curative approach now available for patients who have CML. The disease status at the time of transplantation is the most powerful predictor of DFS. Thus, most centers recommend allo-BMT for any child who has adult-type CML in the chronic phase, preferably within 1 year from diagnosis.
The introduction of STI-571 (Gleevec) as an ABL-specific tyrosine kinase inhibitor will have a significant impact on the natural history, treatment, and survival rates of CML in the near future. As a single agent, it has shown significant activity in patients in the chronic phase who are refractory to interferon177 as well as patients in blast crisis.178 In combination with traditional chemotherapeutic agents, imatinib has shown great promise in a COG trial for treatment of Ph-positive ALL without need for HSCT.179 The optimal regimens and dosage of imatinib, dasatinib, and others are under investigation, but will likely revolutionize treatment for CML.
JMML is a clonal disorder that begins at the pluripotent stem cell level. Although the predominant abnormality is observed in the monocyte, JMML is also associated with disordered erythrocyte, platelet, and lymphocyte function.180 Common clinical features at diagnosis include age less than 2 years, persistent respiratory infection (with tachypnea, cough, wheezing), prominent lymphadenopathy, splenomegaly, skin rash (eczema, xanthomata, and café-au-lait spots), bleeding, and failure to thrive. In addition, anemia, leukocytosis, thrombocytopenia, monocytosis, and nucleated RBCs in the peripheral blood are common laboratory findings.181,182 Cytogenetic analysis may be abnormal (e.g., monosomy 7), but the Philadelphia chromosome is never found. The disease is generally more aggressive than CML with a median survival of 9 months; most patients die of infection.183 Traditional chemotherapy with single agents (e.g., busulfan, hydroxyurea) or intensive multiagent regimens have been unsuccessful. BMT is currently the standard choice of treatment of JMML. Currently, a clinical trial sponsored by the International JMML Registry is testing the efficacy and safety of farnesyl transferase inhibitors for this patient population.
Non-Hodgkin’s Lymphoma
Epidemiology and Etiology
Lymphomas, which constitute about 15% of cancer diagnoses in patients younger than 20 years of age, are the third most common form of childhood cancer. In the United States, approximately 2100 children and adolescents under age 20 years are diagnosed with lymphoma each year.1 Approximately 800 of these are NHL. The incidence peaks between 5 and 15 years of age and is higher in males than in females and in Whites than in African Americans. In younger children, NHL is more frequent than Hodgkin’s lymphoma (HL), whereas in adolescents HL is the more common type of lymphoma. Congenital immunodeficiency syndromes and acquired immunodeficiency syndrome (AIDS) are associated with an increased risk of NHL.184–186 The association of immunosuppressive drug therapy including corticosteroids and androgens with development of malignant lymphomas is well described.186 With the increasing rate of successful organ or marrow transplantation, the number of children with posttransplant lymphoproliferative disease (PTLD) represents an increasingly significant proportion of patients with NHL.187,188 Cases that are seen associated with diseases known to carry an increased risk still represent the minority of NHL diagnoses; overall, the cause is unknown in most cases.
Classification
These lymphomas are a heterogeneous group of malignancies arising from transformation of cells of lymphocytic origin, but are quite variable in terms of clinical manifestations, site of origin, histopathology, immunophenotype, and cytogenetic abnormalities. Pediatric NHL is an aggressive disease with diffuse, high grade, histologic features in which the malignant cells appear undifferentiated or poorly differentiated.189 NHL in children is characterized by disseminated disease at diagnosis, frequent extranodal involvement (including tonsils, thymus, and Peyer patches), and involvement of the bone marrow and the CNS. This is in notable contrast to the NHL seen in adults, where localized, low or intermediate grade, nodal tumors predominate.
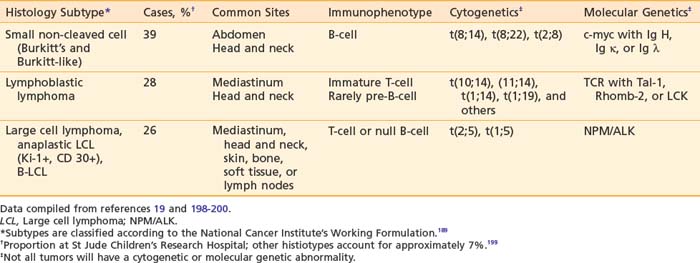
Pediatric NHL can be classified into three major categories based on predominant cell type and architectural pattern: small, noncleaved cell (Burkitt and non-Burkitt), large cell, and lymphoblastic lymphoma. All three histologic subtypes are classified as high grade according to the NCI Working Formulation for Clinical Usage.166 These three major subtypes also can be categorized further according to immunophenotype, cytogenetic abnormalities, and molecular features as shown in Table 54-6.190 Within the small noncleaved cell category are two histologic variants, the typical, uniform starry sky pattern of Burkitt lymphoma and the heterogeneous population of variable sizes designated as Burkitt-like lymphoma. Differentiation of Burkitt-like from diffuse B large cell lymphoma (DLBCL) is difficult based on histology alone and requires significant expertise and whenever possible additional cytogenetic and molecular information. DLBCL, also a mature B-cell neoplasm, presents typically in the adolescent age group with a clinical picture similar to Burkitt and Burkitt-like lymphoma, but rarely affects the bone marrow or CNS. Several chromosomal abnormalities have been shown to be pathognomic for certain subtypes of NHL. For example, the translocation t(8;14) correlates with Burkitt lymphoma191 and the nonrandom chromosomal translocation t(2;5) is expressed by the Ki-1 (CD30) positive anaplastic large cell lymphoma (T-cell immunophenotype).192 These genetic abnormalities are useful primarily in classification but not in predicting outcome or modifying therapy.193 Along with stage of disease, the classification according to histology and immunophenotype is the most important factor in determining appropriate therapy and predicting outcome.194–197 Follicular lymphoma is exceedingly rare in childhood.
Clinical Presentation
The signs and symptoms at diagnosis of NHL in pediatric patients are dependent on the anatomic site and the extent of disease.198–200 The abdomen is the most common site of disease, seen in 35% of cases, and frequently presents with nausea and vomiting; abdominal pain, swelling, or both; change in bowel habits; and ascites. The symptoms may mimic intussusception with bloody diarrhea or appendicitis since a common site of tumor infiltration is the distal ileum, appendix, or large bowel, resulting in bowel obstruction by extrinsic compression or intussusception. Primary involvement with a mediastinal mass occurs in about 25% of cases and may be accompanied by pleural effusions. Symptoms may include chest pain, dyspnea and cough, dysphagia, and swelling of the neck and face (superior vena cava syndrome). The head and neck region, including the Waldeyer lymphoid tissues and cervical lymph nodes, is involved in up to 30% of cases. Presenting features include unilateral tonsillar enlargement and orbital or jaw masses.
The presenting site of disease and thus clinical features also correlate with the histopathologic subtype as shown in Table 54-6.201 The majority of the abdominal lymphomas are small non-cleaved cell type or Burkitt’s lymphoma. In the lymphoblastic lymphomas the typical primary site is the mediastinum. Large cell lymphomas can present with an anterior mediastinal mass, abdominal primary tumor, or head and neck involvement. Anaplastic large cell lymphoma frequently involves the lymph nodes and can involve other atypical sites such as the skin, lung, thyroid, soft tissues, and bone. Relative to the other subtypes of pediatric NHL, marrow and CNS involvement are uncommon. Tumors presenting in the head and neck are seen with any of the four major histologic types.
Staging
The most widely used staging system in childhood NHL is St Jude Children’s Research Hospital Staging System, described by Murphy (Table 54-7).201 It is applicable to all histologic types of childhood lymphoma. For treatment purposes, patients often are stratified into treatment groups based on whether they have limited-stage or advanced-stage disease that corresponds to stages I/II and III/IV, respectively, of the St Jude system.
Table 54-7 The St Jude Staging System for Childhood Non-Hodgkin’s Lymphoma
| Stage | Description |
|---|---|
| I | A single tumor (extranodal) or single anatomic area (nodal), excluding the mediastinum or abdomen |
| II | A single tumor (extranodal) with regional lymph node involvement on same side of the diaphragm: |
| III | On both sides of the diaphragm: |
| IV | Any of the above with initial CNS or bone marrow involvement (<25% blasts) |
From Murphy SB: Classification, staging and end results of treatment of childhood non-Hodgkin’s lymphomas: dissimilarities from lymphomas in adults, Semin Oncol 7:332, 1980.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


