53 Pediatric Central Nervous System Tumors
Epidemiology and Etiology
Pediatric central nervous system (CNS) tumors account for nearly 20% of all childhood neoplasms, exceeded in incidence only by leukemia, and are the leading cause of cancer death in childhood. Incidence is about 1 per 30,000 children each year.1 Although roughly two thirds of all patients are alive 5 years after diagnosis, survivors suffer from neurologic, cognitive, psychologic, and endocrinologic sequelae of both the disease and its treatment. Boys and girls are equally affected, except for medulloblastoma, germ cell tumors, and ependymoma, in which more boys are affected.2–4 Tumors can rarely appear in association with genetic syndromes (Table 53-1), but more often appear to arise spontaneously. The tumors are sometimes found to harbor somatic genetic changes, such as mutations in single genes in atypical teratoid rhabdoid tumor (ATRT; INI1) or high-grade astrocytoma (p53), aberrant gene expression pathways in medulloblastoma (SHH, Wnt, and Notch), and chromosomal losses or gains in high-grade astrocytoma (10), medulloblastoma (17p, 8q24), ependymoma (22), and germ cell tumors (12p).5 Some tumors, do not have a consistent genetic or chromosomal abnormality. Environmental exposures, aside from radiation, have been difficult to associate consistently with development of any CNS tumor. High-dose ionizing radiation clearly increases the risk of developing a meningioma or malignant glioma.6
Table 53-1 Syndromes Associated With Pediatric Central Nervous System Tumors
| Syndrome | Associated CNS Tumors |
|---|---|
| Basal cell nevus (Gorlin) | Medulloblastoma |
| Cowden | Dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos) |
| Li-Fraumeni | Astrocytomas, medulloblastoma, choroid plexus tumors |
| Neurofibromatosis 1 | Pilocytic astrocytoma, malignant peripheral nerve sheath tumor |
| Neurofibromatosis 2 | Schwannoma, meningioma, ependymoma |
| Rhabdoid tumor predisposition | Atypical teratoid rhabdoid tumor |
| Tuberous sclerosis | Subependymal giant cell astrocytoma |
| Turcot | Medulloblastoma, glioblastoma |
| von Hippel-Lindau | Hemangioblastoma |
CNS, Central nervous system.
Anatomy
In contrast to adults, the majority of pediatric CNS tumors are infratentorial (i.e., cerebellum, brain stem, and fourth ventricle) and primary in origin. The location of the tumor may be related to cause; for example, germ cell tumors are likely to develop in the midline periventricular region after abnormal migration of stem cells, and medulloblastoma may arise in the cerebellar external granule cell layer. Tumor location also dictates presenting symptoms (see Clinical Presentations, in the following text). For example, intrasellar or parasellar lesions may produce hormonal and visual abnormalities. Anatomy likewise plays a key role in determination of treatment and prognosis. For instance, pilocytic astrocytomas of the cerebrum or cerebellum are often completely resected and fully cured, whereas those in the diencephalon or brainstem can less easily be resected and require radiotherapy or chemotherapy. Patients with ependymoma, or embryonal and germ cell tumors involving the ventricles, may require more extensive postoperative irradiation as the circulation of the cerebrospinal fluid (CSF) is an important route for tumor dissemination.
Pathologic Conditions
Whereas most adult primary brain tumors are high-grade astrocytomas, the heterogeneity of histologic diagnoses in children is striking. Photomicrographs of the most common tumors, pilocytic astrocytoma, medulloblastoma, ependymoma, and germinoma, are shown in Fig. 53-1 through Fig. 53-4. Historically, brain tumors have been identified by correlative normal cells of putative origin: astrocytes, neurons, and oligodendroglia. However, biologic proof of such derivation is wanting.7 There is only minimal correlation between the incidence of particular histologic types of cancer in the brain and the absolute number of potential precursor cells in that region.8
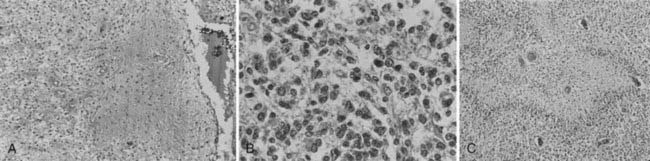
FIGURE 53-1 • Astrocytoma. A, Grade I; B, grade II; C, grade III.
(Courtesy Dr. Richard Davis, University of California–San Francisco.)
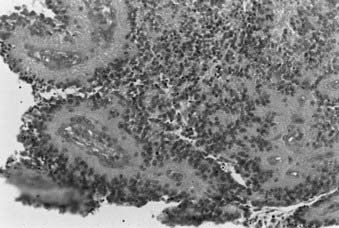
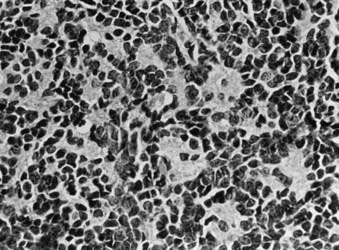
FIGURE 53-3 • Medulloblastoma: Homer-Wright rosettes.
(Courtesy Dr. Richard Davis, University of California–San Francisco.)
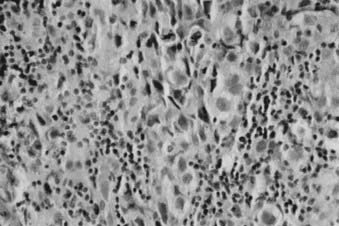
At present, the most recent classification by the World Health Organization (WHO) sorts CNS tumors as follows: tumors of neuroepithelial tissue, tumors of cranial and paraspinal nerves, tumors of the meninges, tumors of the sellar region, lymphomas and hematopoietic neoplasms, germ cell tumors, or metastatic tumors.5 Neuroepithelial tumors can be subtyped further into astrocytic, oligodendroglial, oligoastrocytic, ependymal, choroid plexus, neuronal, pineal, or embryonal tumors. Although the majority of pediatric brain tumors are reported to be glial in origin (e.g., pilocytic and diffuse astrocytomas), children also display a diversity of tumors seldom or never seen in adults: medulloblastoma, choroid plexus papilloma and carcinoma, ganglioglioma, astroblastoma, pleomorphic xanthoastrocytoma (PXA), and dysembryoplastic neuroepithelial tumor (DNT). Previously, medulloblastoma and other embryonal neoplasms were all lumped together as primitive neuroectodermal tumors (PNETs), but now are known to be biologically distinct as medulloblastoma, atypical teratoid rhabdoid tumor, pineoblastoma, ependymoblastoma, and other supratentorial PNETs.9 Certain adult tumors, such as oligodendroglioma, oligoastrocytoma, and well-differentiated astrocytoma, are relatively uncommon in children, and such diagnoses should be questioned for accuracy. For example, DNT is commonly mistaken as oligodendroglioma and PXA sometimes misinterpreted as glioblastoma.10 Calcification and cyst formation are representative of a chronic course and typically indicative of more indolent tumors, such as craniopharyngioma, epidermoid tumor, ganglioglioma, and pilocytic astrocytoma. For the pediatric patient with a CNS tumor, consultation with a neuropathologist skilled in the range of childhood brain tumor diagnoses is highly warranted, especially because diagnosis determines treatment planning.
Tumor grade less often determines treatment and outcome, compared with adults. Although the WHO employs a traditional I to IV scale of increasing anaplasia, some tumors have no variability in grade, or, for some, tumor grade may not distinguish outcome or be mistakenly reassuring of outcome. All medulloblastomas are recorded as grade IV, although there is now a movement to subclassify these neoplasms based on anaplasia, desmoplasia, and nodularity.10 Ependymomas are graded only as well-differentiated (WHO grade II) or anaplastic (WHO grade III), although numerous studies have failed to show a difference in outcome.11 All pilocytic astrocytomas are graded as I. Nevertheless, suprasellar and other midline pilocytic tumors often require chemotherapy or irradiation and result sometimes in death, unlike the grade I cerebral and cerebellar gangliogliomas and pilocytic astrocytomas cured simply by surgical removal. The majority of astrocytomas in children are pilocytic astrocytomas (WHO grade I) and do not undergo anaplastic transformation, in distinction to diffuse astrocytomas (well-differentiated astrocytoma [WHO II], anaplastic astrocytoma [WHO III], and glioblastoma multiforme [WHO IV]), more commonly seen in adults. Recently the diagnosis of pilomyxoid astrocytoma has been adopted by the WHO as a grade II, more aggressive variant of pilocytic astrocytoma.5
Clinical Presentations
The presenting symptoms and signs of intracranial lesions in the developing child vary, depending on tumor location, grade or growth rate, and age at presentation (Table 53-2). These factors also influence the time to diagnosis. A long prodrome over months to years often indicates a more favorable histology and grade, whereas a short prodrome of just weeks or months may indicate an aggressive neoplasm. Symptoms and signs may be generalized (e.g., developmental delay, irritability, growth failure), or secondary to an increase in intracranial pressure (e.g., headache, vomiting, papilledema). If the sutures have not yet fused, toddlers often manifest increased intracranial pressure chronically by macrocephaly with an increased head circumference, and sometimes split cranial sutures or tense, open anterior fontanelle.
Table 53-2 Frequent Symptoms and Signs of Pediatric Central Nervous System Tumors
| Infants | Children |
|---|---|
| Failure to thrive | Headaches |
| Bulging fontanelles | Nausea and vomiting |
| Arrested development | Lethargy |
| Head nodding | Ataxia |
| Nystagmus | Cranial nerve palsies |
| Seizures | |
| Visual field abnormalities |
Routes of Spread
Childhood astrocytomas, other gliomas, and a number of other brain tumors generally do not relapse or recur far from the primary lesion. Unfortunately, as vital structures are compressed within the confines of the skull, it is destruction of the primary organ, rather than metastasis, that results in great morbidity and mortality. Malignancies such as medulloblastomas, ATRT, pineoblastoma, supratentorial PNET, germ cell tumors, and sometimes ependymoma can spread through the ventricular system and metastasize along the meninges or spine. It is particularly common for medulloblastoma to metastasize beyond the posterior fossa to involve the meninges or parenchyma of the spine or cerebrum. Although brain tumors can metastasize outside the CNS, this is currently thought to be a rare occurrence, and the documentation of such events in earlier reports may have been the result of tumor cells that spread through unfiltered shunts during an era when chemotherapy was seldom used for brain tumors.12 Hematogenous and lymphatic tumor spread are not relevant in brain tumors.
Diagnostic and Staging Studies
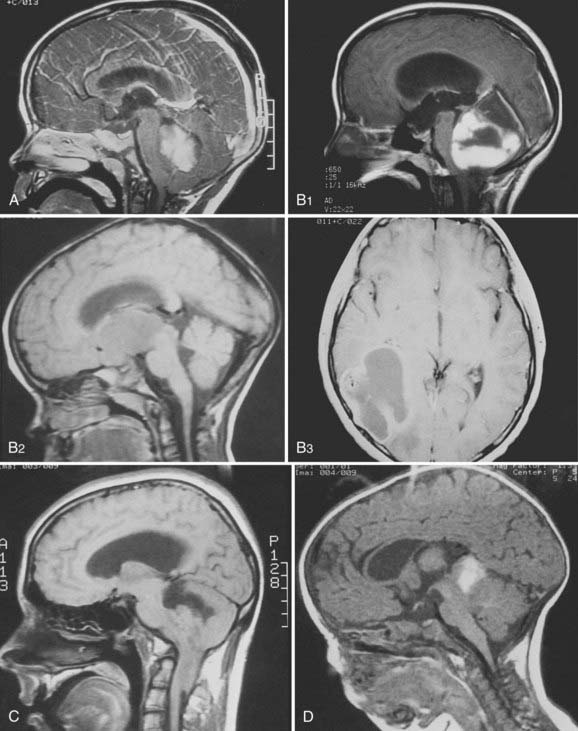
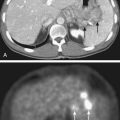
Dramatic improvement in neuroimaging during the past 2 decades have resulted in major advances in diagnosis and targeting of therapy for lesions of the CNS. Young children often require sedation or general anesthesia for any study, but magnetic resonance imaging (MRI) yields the best definition of parenchymal lesions, as well as clear views of the posterior and middle cranial fossa. MRI has replaced myelography for evaluation of the spine, and is therefore the modality of choice for evaluation of the craniospinal axis. Preoperative MRI with gadolinium contrast should include images of the brain, and when there is a posterior fossa tumor or a supratentorial tumor suspected to be embryonal or germ cell, the entire neuraxis should be imaged. Postoperative MRI scan with gadolinium contrast needs to be performed within 48 hours of surgery to evaluate the extent of resection, prior to the onset of postoperative edema and potentially artifactual enhancement. All these MRI data are of critical importance for planning further treatment and for defining the gross tumor volume (GTV) and the clinical treatment volume. Following radiotherapy, the results of treatment are followed by routine interval MRI scans, and in cases of ambiguous findings, MRI techniques such as spectroscopy and diffusion-weighted imaging, or positron emission tomography (PET), may be used to distinguish between recurrent tumor and treatment effects, such as radiation necrosis. Figure 53-5 demonstrates the radiographic appearance of the common tumors by MRI.
Staging System
In contrast to staging systems for other types of cancer, CNS tumors are staged mostly by extent of metastasis using the M scoring of the Chang staging system (Table 53-3), with some consideration of extent of resection.13 Tumor extent at diagnosis by the Chang T score is no longer used. Indeed, T3B brainstem invasion in medulloblastoma was once considered to place a patient at higher risk of recurrence, but has now been shown not to be predictive.14 At present, medulloblastoma in children ages 3 and older is risk-stratified as average- and high-risk. Less than total resection (>1.5 cm2 residual), any metastasis (M+), or severe anaplasia on the histologic specimen place a child into high-risk status. Only complete resection, no metastasis, and absence of diffuse anaplasia are consistent with average risk. For other pediatric CNS tumors, histologic type and M-stage largely indicate risk and dictate dosage and volume of radiotherapy, whereas extent of removal merits therapy based only on the histologic type.
Table 53-3 Chang Staging System
| T1 | Tumor <3 cm |
| T2 | Tumor >3 cm |
| T3A | Extension into the aqueduct of foramen |
| T3B | Invasion of brain stem |
| T4 | Midbrain, third ventricle, or upper cord involved |
| M0 | No metastasis |
| M1 | Microscopic cerebrospinal fluid involvement |
| M2 | Gross seeding of third ventricle |
| M3 | Gross seeding of cord |
| M4 | Extracentral nervous system metastasis |
Standard Therapeutic Approaches
The benefit of performing maximal debulking of the tumor mass safely should be sought, because it improves prognosis and limits adjuvant therapy.15 Only for germ cell tumors has extent of removal been shown not to influence outcome.16 In some instances, surgical resection or biopsy is not performed, namely some optic pathway gliomas (i.e., pilocytic astrocytomas), diffuse intrinsic pontine gliomas, and some germ cell tumors diagnosed by tumor markers. Diffuse intrinsic pontine gliomas are often diagnosed simply by radiographic appearance on MRI of a ventral pontine tumor engulfing the basilar artery in a child with strabismus, ataxia, and weakness over just weeks or several months. Elevated serum or CSF alpha-fetoprotein in the setting of a sellar or pineal tumor is considered diagnostic of an NGGCT, and some individuals consider an elevated serum or CSF beta-human chorionic gonadotropin of less than 50 IU/L without elevation of alpha-fetoprotein diagnostic of germinoma. Except in very young children, incomplete surgical resection of most high-grade malignancies requires postoperative radiotherapy.
Radiotherapy is a key component of treatment for most pediatric CNS tumors. Although planning techniques and the ability to localize tumor volumes have improved significantly in recent years, it is difficult to correlate this progress with improvements in survival. Likewise, it has been difficult to prove any benefit from altered fractionation schemes that have come from radiobiologic studies. In contrast, recent studies have suggested that sophisticated treatment planning and sparing of normal tissues have perhaps mitigated neurocognitive sequelae associated with radiation therapy to the young brain (see Toxicity of Radiation Therapy, in the following text).
Interstitial brachytherapy allows for the delivery of high doses of radiation to a tumor region. One retrospective study reports the largest series of pediatric brain tumors treated with iodine-125 (I-125) brachytherapy.17 Twenty-eight children were treated with temporary, high-activity 125I brachytherapy for recurrent or persistent supratentorial tumors. The study is most useful in its documentation of acute and late toxicities. No grade 3 or grade 4 acute or late toxicities occurred. However, 22 patients (79% of 28 total patients) required at least one repeat operation following brachytherapy, and 17 of these 22 patients had evidence of necrosis in the resected specimen. Furthermore, little improvement was found compared with conventional methods.
To date, outcome data with use of proton beam radiotherapy for CNS tumors is limited, and advocated mostly on theoretical grounds. Merchant and colleagues compared models of photon and proton dose characteristics and their predicted relationship to cognitive function in children treated for brain tumors.18 They found that small critical normal structures such as the cochlea and hypothalamus, which were anatomically separated from the PTV, received substantially less radiation using protons compared with photons. Proton radiotherapy would therefore be expected to reduce the risks of endocrine deficits and hearing loss. In addition, protons decreased the low (0-20 Gy) and intermediate (20-40 Gy) doses to the cerebrum in patients receiving focal radiation. Using longitudinal models of radiation dose-cognitive effects, their data indicate that proton radiotherapy would mitigate intellectual decline from radiation treatment.
The benefit of proton beam radiotherapy in children may be best exemplified by its use for craniospinal irradiation (CSI). Comparisons of proton beam, conventional 3-D radiation, and IMRT for treatment of the posterior fossa and spinal column suggest superior sparing of normal structures by protons. In particular, protons are likely to mitigate long-term toxicities related to hearing, endocrine, and cardiac functions. For example, 90% of the cochlea received 101.2% of posterior fossa boost dose with conventional radiation techniques, 33.4% with IMRT, and only 2.4% with protons. Similarly, 50% of the heart received 72.2%, 29.5%, and 0.5% of the posterior fossa boost dose for conventional x-ray, IMRT, and protons, respectively.19
Toxicity of Radiation Therapy
A wide range of potential toxicities complicates the implementation of radiation therapy in the treatment of tumors of the craniospinal axis. These toxicities can be severe and debilitating, particularly in pediatric patients. Care should be taken to minimize these effects. Treatment of intracranial tumors can result in damage to the eye, ear structures, brain, and hypothalamic-pituitary axis, as well as growth abnormalities. Treatment of the spine can result in growth deficits and damage to the spinal cord. Specific potential acute side effects of radiation to the CNS include epilation, skin reactions, otitis, hematopoietic depression, and somnolence. Specific late toxicities of such radiation include radionecrosis, myelopathy, leukoencephalopathy, vascular injury, neuropsychologic sequelae, endocrine dysfunction, bone and tooth abnormalities, ocular complications, ototoxicity, and induction of second primary tumors. Table 53-4 delineates the radiation doses associated with late toxicities that may result from radiation therapy to the CNS.
Table 53-4 Late Toxicities of Central Nervous System Irradiation
| Structure | Late Effect | Threshold Dose |
|---|---|---|
| Spinal cord | Chronic progressive myelitis | 45 Gy |
| Brain | Radiation necrosis Intellectual deficits | 54 Gy 12-18 Gy |
| Lens of eye | Cataract formation | 8 Gy |
| Retina | Radiation retinopathy | 45 Gy |
| Optic nerve | Optic neuritis | 50 Gy |
| Inner ear | Sensorineural hearing loss | 40-50 Gy |
Spinal Cord
Radiation therapy can result in severe damage to the spinal cord, with functional transection of the cord at the affected level representing the most dire of such toxicities. Radiation damage takes the form of chronic progressive myelitis. The 5% incidence of radiation myelopathy probably lies between 57 Gy and 61 Gy in the absence of chemotherapy, using standard fractionation. At doses less than 45 Gy the risk of myelitis is less than 0.2%. At 73 Gy the risk of myelitis approaches 50%.20,21 A number of reports have indicated that tolerance of the cervical spinal cord to radiation toxicity is somewhat higher than 45 Gy in adults. However, it is unclear what the cervical spinal cord tolerance is in pediatric patients.
Brain
Acute reactions during radiation therapy, thought to result from disruption of the blood-brain barrier, are uncommon. However, there are reports of edema following single conventional fractions.22 Clinically, apparent acute changes are more common with hypofractionated doses, such as are used in radiosurgery.23 Subacute reactions are more common and are thought to be due to transient demyelination.24 These effects typically occur within the first few months following radiation and usually resolve within 6 to 9 months. Cranial nerve palsies have also been reported.25 Severe subacute effects such as rapidly progressive ataxia are rarer and are generally associated with fractions larger than 2.0 Gy and total doses larger than 50 Gy.26
The late effects of radiation are primarily due to radiation necrosis. Symptoms are related to the neuroanatomic location of necrosis (sensory, motor, speech/receptive deficits, seizures) and may, in addition, be due to increased intracranial pressure. Focal necrosis is uncommon with doses less than 60 Gy given with conventional fractionation.27
Large-volume radiation therapy can result in diffuse white matter changes. Clinically, these can result in lassitude, personality change, or neurocognitive deficits. Multiple studies have examined the effect of whole-brain radiation therapy on intellect in patients treated for leukemia. Radiation-associated depression in intelligence quotient (IQ) has been noted by a number of authors.28,29 Halberg and colleagues30 compared three groups of patients. The first group received 18 Gy (1.8 Gy per daily fraction) cranial irradiation, and the second received 24 Gy cranial irradiation. The third group consisted of other oncology patients who did not receive cranial irradiation. Lower IQ scores were noted in the group receiving 24 Gy. High-dose methotrexate and female sex appear to increase risk of intellectual deficits.31,32 The effects of cranial irradiation are also more severe the younger the child. Intellectual deficits resulting from radiation most commonly result in difficulty acquiring new knowledge, decreased processing speed, and memory deficits (most frequently short-term).33
Pediatric CNS tumors require a multimodality approach, including surgery, chemotherapy, and radiation. Attribution of toxicities such as neurocognitive decline to a single modality, most often radiotherapy, is not supported by the literature. Kiehna and colleagues34 conducted a prospective trial of 120 children and young adults to evaluate neurocognitive aptitudes before, during, and after CRT. In this comprehensive study from St. Jude Children’s Research Hospital, investigators used the Conners continuous performance test to measure selective attention, sustained attention, impulsivity, and reaction time. During a 6- to 7-week course of CRT, children exhibited problems with cognitive processing, attention, and reaction times. Overall attention indices worsened during CRT, with significant risk factors including progression of symptoms prior to CRT, young age, and diagnoses of craniopharyngioma or low-grade astrocytoma.34
The group at St. Jude Children’s Research Hospital has also evaluated academic achievement and verbal memory performance in children after CRT. Factors including female gender, number of surgeries, and hydrocephalus, predicted worse verbal memory performance at baseline. Of key importance, however, verbal memory stabilized and improved over time, rather than declined.35 In a similar study from the same group, serial assessments of academic achievement revealed that reading was the aptitude most susceptible to decline after CRT, whereas math and spelling skills remained stable.36
Ear
Radiation-induced sensorineural hearing loss is dose-dependent and is more severe in younger patients. Hearing loss can result from doses greater than 40 to 50 Gy, usually developing within 6 to 12 months of treatment.37 High-frequency hearing loss is seen in 25% to 50% of patients who received greater than 50 to 60 Gy to inner ear structures.38–40 Hearing loss is typically attributed to radiation changes induced in the cochlea and vasculature. Ototoxicity related to cisplatinum chemotherapy is well documented.41,42 Cranial irradiation before or concurrent with cisplatinum chemotherapy enhances ototoxicity.41,43 Chronic otitis can also develop following radiation therapy because of obstruction of the eustachian canal.
Endocrine
Whole-cranial irradiation or more focal radiation that includes the hypothalamic-pituitary axis can result in neuroendocrine abnormalities. The neuroendocrine complications that can result from radiation therapy are summarized in Table 53-5. Growth hormone (GH) production appears to be the most prone to disruption by radiation therapy. GH deficiency worsens over time and may follow radiation doses as low as 12 Gy.44

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


