55 Pediatric Bone and Soft Tissue Tumors
Osteosarcoma and Ewing sarcoma are first and second in incidence of childhood bone tumors and compose 2.6% and 2.1% of pediatric solid tumors (approximately 260 and 200 cases annually), respectively.1 Most primary osteosarcoma lesions are managed by amputation or limb-sparing procedures and intensive chemotherapy. Pulmonary metastases are managed by surgical excision. For primary lesions in an unresectable site, control with conventional photon radiotherapy is limited. With proton-beam therapy, 60% local control was achieved for patients with axial skeletal tumors.2 Hypofractionated, accelerated radiotherapy afforded durable response in 14 patients requiring palliation of primary or metastatic lesions.3 For 17 patients with spine osteosarcoma, radiotherapy after intralesional surgery improved survival.4 A dose escalation trial of 153-samarium-ethylenediaminetetramethylphosphonic acid followed by peripheral stem cell rescue demonstrated minimal toxicity and prolonged response in patients with recurrent or metastatic osteosarcoma.5
Other rare bone tumors may present in childhood. Non-Hodgkin lymphoma of bone responds well to multiagent chemotherapy, and radiotherapy is required only for nonresponders.6 Malignant fibrous histiocytoma of bone can be controlled with radiotherapy.7 Hemangioendothelial sarcoma of bone has a variable presentation and natural history and may be multifocal in one limb or bone.8 Radiotherapy to 40 to 60 Gy affords good local control.8–11 Benign giant cell tumor of bone presents in the pediatric age group in locations not amenable to surgery.12 Radiation to greater than 40 Gy resulted in a local control rate of 75% to 85% in two series.13,14 Proton-beam therapy has also been used with similar control rates.2 Local control of aneurysmal bone cyst in three adolescents was achieved with 40 Gy.15
Ewing Sarcoma of Bone
Epidemiology and Cytogenetics
A case-control study of environmental and familial factors did not identify risk factors for Ewing sarcoma.16 A reciprocal translocation t(11;22)(q24;q12) characterizes Ewing sarcoma of bone and is shared with extraosseous Ewing sarcoma and peripheral primitive neuroectodermal tumor (of bone or soft tissue), and helps define the Ewing sarcoma family of tumors.17,18 Mapping of the translocation break point indicates fusion of the FLI-1 gene on 11q24 with the EWS gene of 22q12.19 One function of the resulting fusion protein is repression of a tumor suppressor gene that encodes for the type II receptor of transforming growth factor beta.20 Patients harboring the less frequent EWS-ERG fusion protein resulting from a t(21;22)(q22; q12) translocation were compared with patients with the EWS-FLI1 transcript. Differences in clinical phenotype and outcome were not observed.21
As transcriptional repression by NKX2.2 is mediated through histone deacetylases (HDACs), these findings identify a novel therapeutic approach for Ewing sarcoma. Specifically, HDAC inhibitors may prove clinically effective by reversing NKX2.2-mediated transcriptional repression. This line of investigation exemplified how scientific investigations can indicate promising novel approaches to cancer treatment.22
Clinical Presentation and Routes of Spread
Ewing sarcoma of bone may present as late as the fourth and fifth decades of life, but the mean and median age (15 to 16 years) reflect the predominance of presentation between ages 10 and 20 years (approximately 65% of cases).23–25 Age at diagnosis younger than 5 years is rare, as is diagnosis in blacks. The male/female ratio is 1.5 : 1. Pain is the most common presenting symptom (>90%), and two-thirds of patients complain of a mass lesion. When a long bone is the primary site, the tumor is typically in the metaphysis or diaphysis. Three-fourths of primary sites are extremities or pelvic bones, with the femur as the single bone most frequently involved (Fig. 55-1).26 Other features at presentation include pathologic fracture (16%), systemic symptoms including fever (10% to 30%), and metastases (10% to 25%).23 Of 122 patients in the Intergroup Ewing Sarcoma Studies (IESS-I and IESS-II) with metastases at diagnosis, 53% had lung metastases with or without other metastases and 43% had bone metastases with or without other metastases.27
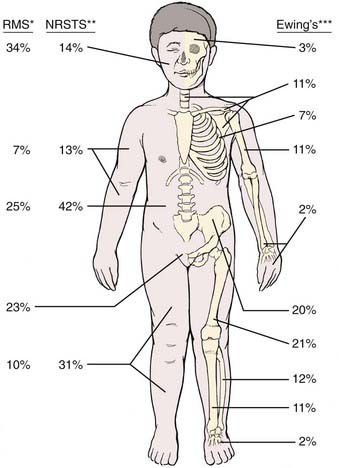
FIGURE 55-1 • Distribution of primary sites for rhabdomyosarcoma (RMS),80 nonrhabdomyosarcoma soft tissue sarcoma (NRSTS),165–167 and Ewing sarcoma.265 *For RMS, the head and neck site may be subdivided as 8% orbit, 8% other head, and 18% parameningeal.The pelvic sites may be subdivided as 11% bladder and prostate, and 12% female genital or paratestis. **For NRSTS, the trunk data include pelvic sites. ***For Ewing sarcoma, certain sites by subdivision are 5% vertebral column; 4% scapula; 12% ilium; and 8% sacrum, coccyx, and pubis.
Pathologic Conditions
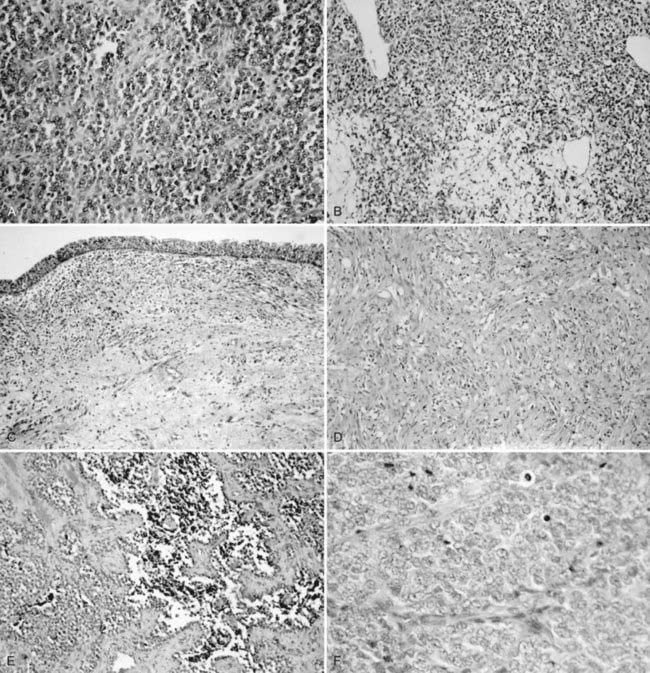
The histogenesis of Ewing sarcoma is uncertain. The classic or “typical” variant is shown in Fig. 55-2A. A variant featuring greater range of cell size and more frequent mitoses has been termed atypical and may also have a lobular, alveolar, or organoid organizational pattern. The primitive round-cell sarcoma of bone is less common and on light microscopy may have cells of greater size and shape, more pink cytoplasm, and a more prominent matrix. Ultrastructural features are distinct from Ewing sarcoma of bone and include the presence of cytoplasmic organelles, intracytoplasmic attachments, and developed intercellular attachments. Neurosecretory granules are not apparent in contrast to the peripheral primitive neuroectodermal tumor.28 There is at present insufficient clinical experience to determine if the subtypes differ by age at diagnosis, site of presentation, probability of metastases, and response to therapy.
Diagnostic and Staging Studies
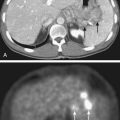
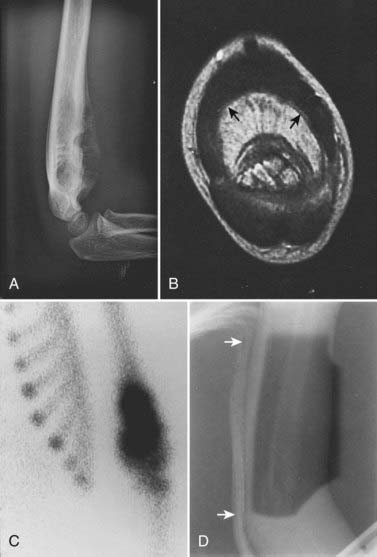
Laboratory studies at presentation, in addition to routine hematology and chemistry, should include serum lactase dehydrogenase level and histologic assessment of bone marrow. Chest computed tomography (CT) and bone scan are required. Imaging studies of the primary lesion include plain radiographs that typically show a central destructive lesion with variable sclerosis and osteolysis. Periosteal elevation by the tumor mass stimulates calcium deposition that may form layers with an “onion skin” appearance and that may, at the periphery of the elevation, demonstrate the Codman triangle sign (Fig. 55-3). A soft tissue component may show radiating calcium spicules. Bone scan will also show the extent of the primary lesion.29 Although CT assessment of the primary site is superior to magnetic resonance imaging (MRI) for identification of tumor matrix calcification and periosteal reaction, MRI has several advantages. The assessment of bone cortex is best seen in the axial plane. Sagittal and coronal images show (on T1 sequences) the extent of marrow infiltration. Major blood vessels are well seen. Soft tissue tumor mass and adjacent normal muscle are distinguished on T2-weighted images.30 The coronal and sagittal images are best suited for determining the gross tumor volume (GTV), but correlation with the bone scan is mandatory.
After treatment, an elevated serum lactase dehydrogenase level may herald relapse.31 Plain radiographs, CT, and MRI of the primary lesion will show primary site recurrence.30,32 The bone scan should return to normal within a median of 12 months (range 6 to 18 months) after radiotherapy. Recurrence of uptake at a median of 10 months after radiation (range 6 to 12 months) or persistent uptake signals primary site tumor regrowth or, occasionally, pathologic fracture or ectopic bone formation.29
Staging System
No staging system is in common use for Ewing sarcoma of bone. Pretreatment tumor characteristics that portend a poor prognosis include presence of metastases,23 proximal or central sites,33 volume greater than either 100 or 500 ml,33,34 and gross soft tissue extension.35
Standard Treatment
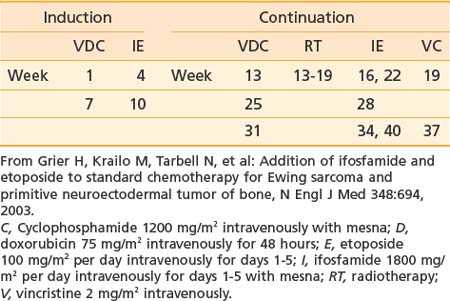
Treatment of Ewing sarcoma requires prompt initiation of chemotherapy at completion of diagnostic evaluation. Treatment of the primary site with radiotherapy, surgery, or both follows induction chemotherapy. Intergroup Study 0091 demonstrated the superiority of a six-drug regimen.36 A modified regimen for patients receiving definitive radiotherapy on the subsequent Children’s Oncology Group (COG) Study AEWS 0031 is shown in Table 55-1 and represents a contemporary standard.
The propensity for Ewing sarcoma to extend within the medullary cavity or into adjacent soft tissues has limited the application of limb-sparing procedures successfully used for osteosarcoma patients. Historically, surgical resection was restricted to “expendable” bones (e.g., rib, clavicle, proximal fibula, scapula). The apparent benefit for the surgical approach in retrospective studies (in comparison with radiotherapy) resulted, in part, from selection bias, for which there was no reliable control.37 The proportion of nonmetastatic patients having surgery alone in Intergroup Study 0091 was 38%, with 23% having surgery and radiotherapy and 39% having radiotherapy alone. In the first two European Cooperative Ewing Sarcoma Studies (CESS 81 and 86), the proportion of patients having surgery with or without postoperative radiotherapy was 66%, and the number having preoperative radiotherapy was 5%. In the subsequent European Intergroup Cooperative Ewing Sarcoma Study (EICESS 92), the proportions were 37% and 42%, respectively.38
Role of Radiotherapy
Radiotherapy is used pre- or postoperatively or definitively for the primary tumor and for treatment of pulmonary and skeletal metastases. Management of the primary tumor follows induction chemotherapy. The indications for surgery versus radiotherapy as initial local therapy are still evolving. For the very young child who would have significant leg-length discrepancy after definitive radiotherapy, amputation and prosthetic replacement may be preferred. Smaller lesions in expendable bones should also be considered for primary surgery, as should very large lesions (e.g., in the iliac wing). Sites with pathologic fracture can have definitive radiotherapy if healing occurs during induction chemotherapy; otherwise, surgery is required. After primary surgery, adjuvant radiotherapy is indicated for intralesional resection or positive or close margins. It must be emphasized that the surgical intent should be to completely resect the tumor with negative margins because “debulking” is not an appropriate approach. Although recommended margins of resection are 2 to 5 cm, COG studies define minimal acceptable margins as at least 1 cm for bony margin, at least 5 mm for fat or muscle planes, and at least 2 mm for facial planes. If such minimal resection margins are achieved, postoperative radiotherapy is not indicated. In the three CESS/EICESS trials, local failure occurred in 20% of patients with intralesional resection who had postoperative radiotherapy. The same study showed a less than 5% local failure risk for patients who had good histologic response to initial chemotherapy followed by wide resection (with or without postoperative radiotherapy). The patients given preoperative radiotherapy had a local failure risk of 5%.38
Controversy persists regarding whether surgery or radiation affords greatest likelihood of local control. This debate is particularly pertinent for primary sites such as the pelvis for which surgery often entails extirpative resections. Although no randomized trial has compared local control modalities and no such study is likely to transpire, retrospective studies suggest that local recurrence rates after surgery are lower than those following definitive radiation. However, selection factors that portend worse outcome clearly bias such studies in favor of surgery because larger tumors with poor response to therapy involving sites less amenable to surgery are referred more often for definite radiation. Selection bias notwithstanding, data presented in abstract form analyzes local control modalities in the patients with nonmetastatic Ewing sarcoma of the bone treated on Intergroup Study-0091.36 A total of 329 patients were included, with 122 receiving surgery, 142 definitive radiation, and 65 both local control modalities. At 5 years, cumulative incidences of disease progression were 22.1% for surgery, 36.9% for radiation, and 48.1% for combined surgery and radiation. The investigators used definitive radiation as the reference group and reported that the hazard ratio for local failure was 0.38 (95% CI 0.15-0.98; P = 0.04) for surgery and 0.77 (95% CI 0.34-1.75) for surgery plus radiation and the hazard ratios for death for these two comparisons were 0.77 (95% CI 0.44-1.34) and 1.46 (95% CI 0.88-2.42), respectively. Although confounding factors introduce clear bias in favor of surgery and results therefore must be interpreted with circumspection, rates of disease progression, distant failure, and death did not differ between the surgery and radiation groups. However, patients undergoing surgery had lower local failure rates than those receiving definitive radiation.
The contribution of confounding factors to these conclusions is highlighted by a separate analysis of the same Intergroup Study. In this published report, local control modalities were compared for patients with pelvic Ewing sarcoma enrolled on INT-0091. Seventy-five patients were randomized to receive doxorubicin, vincristine, cyclophosphamide, and dactinomycin (VACA) or VACA alternating with ifosfamide and etoposide (VACA-IE). Neither tumor size nor local control modality affected event-free survival (EFS) or risk of local failure. Cumulative incidence of local failure was 25% for the 12 patients who had surgery, 25% for the 44 who had definitive radiation, and 10.5% for the 19 who had both (P = 0.46).39 In contrast, systemic therapy did influence the risk of local failure. The 5-year cumulative incidence of local failure was 30% for those receiving VACA and only 11% for the patients treated with VACA-IE (P = 0.06), a difference that was evident regardless of local control modality.40
Simulation
Ewing sarcoma can occur in virtually any bone, and therefore the principles of simulation and treatment planning are site-dependent. The initial decision for treatment planning is determination of optimal patient position. For an extremity lesion, a typical plan employs parallel opposed portals and the limb must be positioned so that the lesion, its adjacent soft tissue extension, and the biopsy or surgical scar are all encompassed while leaving a strip of soft tissue outside the port. The avoidance of circumferential irradiation facilitates lymphatic drainage and prevents the late appearance of distal edema. A combination of limb rotation and beam angulation (gantry rotation) usually permits the necessary soft tissue sparing for lesions that have mostly anterior or posterior extension. For the patient with an iliac or sacral primary lesion, the prone position, with or without the use of the “belly board” (which allows bowel to fall forward) will help reduce bowel dose. Bladder distention helps keep the intestine away from the treated volume in patients with a pubic or ischial primary tumor. The patient with a scapular primary tumor may be positioned prone with the involved limb extending off the treatment couch and moved cephalad to place the scapula in a plane that accommodates opposed tangential ports and that maximally spares underlying lung. Immobilization of the treated limb should be done using plaster of Paris or commercially available thermal-reaction polymers. Figure 55-3 illustrates the imaging studies of a child with a primary tumor of the distal left humerus that had anterior soft tissue extension.
Portal margins are based on prechemotherapy imaging studies. Particular attention must be paid to soft tissue extension in axial planes and intramedullary extension of long bones. The contribution of both MRI and bone scan imaging is essential. The typical presentation of the primary lesion in a long bone usually requires inclusion of the adjacent epiphysis in the port. Historically, the volume extended to include the opposite epiphysis, but this is no longer required.41 For the COG study AEWS 0031, the margin requirements were clinical target volume (CTV) equivalent to GTV plus 1.5 cm and planning target volume (PTV) equivalent to CTV plus 0.5 to 1 cm. A reduced portal (CTV = 1 cm) is based on the postinduction chemotherapy, preradiotherapy imaging if there has been regression of soft tissue extension. The reduced portal maintains the same margin around the tumor extent in bone, but is planned to reflect the regression of the soft tissue extension. This is done after a tumor dose of 45 Gy. Exceptions may be made to the rule that the CTV includes soft tissue extension as it existed at the time of diagnosis. Examples include margins where the medial surface of primary lesions in the ileum or rib have displaced bowel or lung (respectively), which then returned to a more anatomic location with regression. The same concept applies for low pelvic lesions that displace the bladder at the time of diagnosis, but do so to a lesser extent after induction chemotherapy. After resection of a primary lesion with microscopic residual, the margins are the original GTV plus 2 to 2.5 cm to 45 Gy; thereafter, a reduced port covers the area of residual tumor (CTV plus PTV = 1.5 to 2 cm) to an additional 5.4 Gy (three fractions).
Radiation Treatment Plan
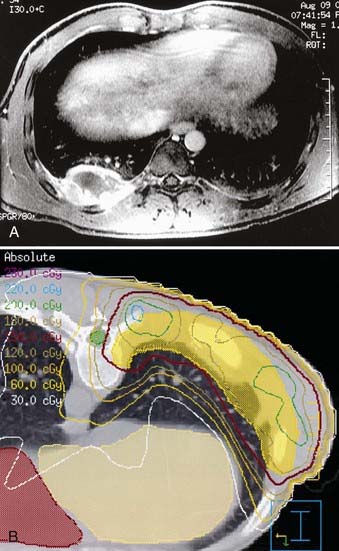
If radiotherapy is given after resection of the primary lesion, the total dose may be limited to 50.4 Gy for the patient with positive margins. Gross residual tumor requires full dose (55.8 Gy). Single institution reports, however, suggest that the postoperative total dose may be reduced to 30 Gy.42 For the patient with a rib primary tumor who has risk factors for pleural involvement, treatment should encompass the entire, ipsilateral hemithorax (1.5 Gy/day to 15 to 18 Gy) before reducing the port to a CTV appropriate to the rib primary tumor (Fig. 55-4).43
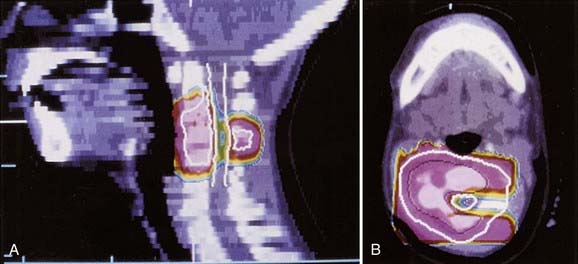
Critical Normal Tissues
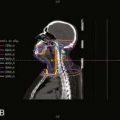
Ewing sarcoma can occur adjacent to virtually every important normal tissue. Fig. 55-5 illustrates the treatment plan for a primary lesion in the third cervical vertebra using proton-beam therapy to minimize dose to the spinal cord. When treating an extremity, the plan should spare a strip of normal tissue. The epiphyseal growth plate opposite the primary tumor can usually be excluded from treatment. Inclusion of only half the growth plate may lead to an exaggerated angle of the affected limb to its adjacent bone. Radiation-induced limb shortening is a function of age at diagnosis and radiotherapy dose. In a study from the orthovoltage era, growth loss for children receiving approximately 50 Gy was about 25% of the remaining (incremental) growth in the unaffected limb.44 Leg dysfunction in patients treated at the National Cancer Institute (NCI) was scored as minor or moderate in 18 of 22 patients. One patient required amputation.45 Late functional results from a study of patients with primary tumors of an extremity who received hyperfractionated radiotherapy (1.2 Gy twice daily; total of 50.4 to 60 Gy) were very good and were subjectively improved over standard fractionation. No pathologic fractures occurred.46 Five-year survivors of lower extremity osteosarcoma and Ewing sarcoma were assessed for limb function and quality of life. Children treated after age 12 and females fared less well. No difference was observed between those having amputation or a limb-sparing procedure.47
Reports of radiation-induced osteosarcomas usually describe patients whose tumor doses were 60 Gy or more. A multi-institutional report demonstrated no secondary sarcomas in patients receiving 48 Gy or less.48 The NCI Surveillance, Epidemiology, and End Results Program included 595 patients with Ewing sarcoma. One osteosarcoma and three soft tissue sarcomas developed in radiotherapy ports.49 Secondary sarcomas developed in 3 of 674 patients enrolled in the CESS trials.50
After radiotherapy, widely distributed lytic changes of radiation osteitis will be seen on the plain radiograph in the initial 2 to 3 years. A localized area of lysis may herald recurrence of Ewing sarcoma or, if at least 5 years have elapsed, a treatment-related bone sarcoma.51
Outcome
Several American institutions have reported treatment results for large patient numbers with 5- and 10-year follow-up. The Mayo Clinic report includes 105 patients with nonmetastatic disease treated from 1969 to 1982. Five-year survival was 42%, and local control was maintained in 73% of patients.23 The NCI report included 80 patients with nonmetastatic disease treated between 1968 and 1980. The 5- and 10-year survival rates were 51% and 39%, respectively. Local control was 80%.25 The IESS-I trial enrolled 342 patients with nonmetastatic disease from 1973 to 1978 in a randomized trial of three treatment regimens. The 5- and 10-year survival rates for the best arm of the study were 65% and 60%, respectively. Local control was 85% and did not differ significantly by treatment assignment.56 The failure pattern for nonmetastatic patients on IESS 0091 was 5% for local only (experimental arm), 20% for systemic (standard or experimental arm), and 2% to 5% for local and systemic or unknown (both arms).36 The EICESS 92 trial enrolled 479 nonmetastatic patients. The 5-year relapse-free survival rate was 59%.53
Several patient and tumor characteristics affect outcome. Three that are interrelated are site, size, and extraosseous extension. Many studies indicate that pelvic, sacral, and other central sites have a worse prognosis. These lesions are typically larger than those at other sites and are more likely to have extraosseous extension. In IESS-I, patients with nonpelvic primary tumors had an overall 5-year survival rate of 57% and, on the superior treatment regimen, 72%. The 5-year survival for patients with a pelvic primary lesion was 34% (no difference by treatment arm), and relapses continued after 5 years. The survival rate was statistically significantly higher in IESS-II for patients with pelvic primary tumors, 63% at 5 years, and the local failure declined from 28% to 12%. The improvement was attributed to more intensive chemotherapy, more aggressive surgery, and the effect of CT on radiation treatment planning.54 For patients with nonpelvic primary lesions, the IESS-II study (1978-1982) compared a regimen similar to the best arm of IESS-I (control arm) with a more intensive schedule of the same drugs. Five-year survival in the intensive arm was 77% and in the control arm was 63% (P = 0.05).55 (The comparable figure for the best arm of IESS-I was 72%, as noted earlier.52,55) The local failure for IESS-II patients with nonpelvic disease was 9%. No site group (proximal, distal, rib, or other) was prognostic for an effect on survival.55
In another report, the presence of soft tissue extension in patients with localized disease was more predictive of failure than large tumor size.35 Significant prognostic factors (endpoint: 10-year EFS) in CESS 86 included tumor size greater than 200 ml (EFS 36%) and less than 200 ml (EFS 63%), as well as poor histologic response (poor response EFS 38% versus good response EFS 64%).56 To assess the relationship of these two variables, patients from the CESS and French EW88 trials were combined and the analysis restricted to 385 nonmetastatic patients who had initial chemotherapy followed by surgery (with or without postoperative radiotherapy). Histologic response (which was independent of tumor size) became the only significant prognostic factor. Based on the percentage of viable cells in the surgical specimen, three prognostic groups were determined (endpoint: 5-year EFS): low risk (<10% cells), 73% EFS; intermediate risk (10% to 49% cells), 56% EFS; high risk (>50% cells), 37% EFS.57 Patients younger than age 3 constituted only 2.6% of the patients enrolled on five IESS protocols. Their 5-year survival rate was identical to that of older patients.58
Outcome of therapy at various uncommon sites has been published: rib, survival rate 50% to 60%43,59; vertebral column, 5-year EFS 36% to 44%60,61; small bones of the hands and feet, 6 of 7 alive; calcaneus, 1 of 5 alive62; and bones of the head and neck, survival rate 80% (see Fig. 55-5).63
Patients with metastases at diagnosis do not have a uniformly fatal outcome. The CESS trials enrolled 114 patients with metastases confined to the lung and pleura. The 10-year EFS for bilateral metastases was 24%; unilateral was 53%. Survival was better for patients who received whole-lung irradiation.64 When patients with skeletal metastases were included, the 4-year EFS was 27%.65 Bone marrow ablative therapy (including total-body irradiation) for patients with bone and marrow metastases did not improve outcome compared with conventional therapy in a report from the Memorial Sloan-Kettering Cancer Center.66
Soft Tissue Sarcoma
Epidemiology
Soft tissue sarcomas in children have an annual incidence of about 650 cases, or 6% of the approximate total of 10,500 new cancer cases per year in U.S. children younger than age 15.1 They therefore rank among pediatric solid tumors after central nervous system and neuroblastoma, before bone tumors and retinoblastoma, and at about the same incidence as Wilms tumor. Slightly more than half of patients have rhabdomyosarcoma (RMS). The remainder, non-RMS soft tissue sarcomas (NRSTSs), are a conglomerate of many types of heterogeneous origin that are individually rare. For both types, the male/female ratio is about 1.2 : 1 and the incidence in both blacks and whites is 8 to 9 per million.67
Causal Factors and Genetics of Rhabdomyosarcoma
An environmental factors study of 322 children enrolled in Intergroup RMS Study (IRS)-III found that parental use of marijuana and cocaine in the year before a child’s birth was highly correlated with the subsequent diagnosis of RMS. The risk was greater (fivefold) for maternal use of cocaine.68
The normal development from uncommitted mesenchymal cell to myoblast (myogenesis) and the cell’s aberrant transformation to malignancy are being elucidated. Myogenic transcription factors are a group of DNA-binding proteins that bind to promoter regions for muscle-specific genes and promote their expression. Several of these factors (MyoD, Myf5, myogenin) are variably expressed in RMS and therefore help to identify a small, blue, round cell tumor without characteristic phenotypic features as RMS and not as another sarcoma such as extraosseous Ewing sarcoma. Defects in the normal pathway are associated with the two major histologic subtypes of RMS: embryonal and alveolar. The embryonal type correlates with loss of heterozygosity at chromosome 11p15.5, with presumed loss of a tumor suppressor locus.69,70 The alveolar subtype features a translocation t(2;13) (q35;q14) that results in the fusion of normally unassociated transcription factors (PAX 3) and the activation domain of FKHR. A variant translocation that produces the PAX 7-FKHR fusion transcript is associated with a better outcome.71 A study of 45 tissue samples of RMS using comparative genomic hybridization and fluorescence in situ hybridization methods demonstrated that genomic gains and losses were similar in alveolar and embryonal histologies, and that they did not differ between the two alveolar fusion transcript subtypes. Both histologies had a 1 : 4 rate of genomic amplification, which, for the embryonal subtype, was most common in those with anaplastic features. One locus of amplification included the gene for insulin-like growth factor type 1 receptor, which has been implicated in the induction of RMS.72
The cancer family syndrome of Li-Fraumeni includes childhood RMS and NRSTS and is characterized by p53 suppressor gene germline mutations.73
Pathologic Conditions
An international committee of pathologists expert in RMS reviewed the histologies of 800 randomly chosen cases from IRS-II.74 The resulting classification retains the basic division between the embryonal and alveolar subtypes, omits the formerly used pleomorphic subtype, and further refines the embryonal category as follows: favorable prognosis, botryoid embryonal and spindle cell (SC) embryonal; intermediate prognosis, all other embryonal; and unfavorable prognosis, all alveolar subtypes (includes cases with any alveolar component) and undifferentiated sarcoma. Examples are shown in Figs. 55-2B through F.
There are several anatomic site associations for the histologic types of RMS. The botryoid tumor occurs in the bladder and vagina, usually in infants and younger children, and 31% of SC variant cases occur in the paratesticular site. Other sites are head and neck, orbit, and extremity.75 About half of patients have an embryonal type that has no particular propensity for specific anatomic sites. Alveolar RMS predominates in extremity and truncal sites. A rare, unfavorable variant containing anaplastic cells is found most often in lower extremity, retroperitoneum and pelvis, and paratesticular sites.76 Whether histologic features are an independent prognostic factor is uncertain. The IRS staging system uses primary site in preference to histologic findings; however, all nonmetastatic patients with alveolar histologic findings were assigned to the intermediate risk trial of IRS-V. The distribution of histologic types on IRS-IV for nonmetastatic patients was 70% embryonal, 20% alveolar, 4% undifferentiated sarcoma, and 6% other.77
Clinical Presentation
Males with RMS predominate in the ratio of 3 : 2, and in IRS-I the race distribution was 80% white, 12% black, and 8% other.78 Median age at presentation is 5 years; and the distribution on IRS-III was 6% less than 1 year and 66% less than 10 years.79 The proportion of all patients presenting with dissemination is 17%.80
Symptoms at presentation relate to anatomic site of the primary tumor. Primary site distribution is shown in Fig. 55-1. The parameningeal site is composed of four sites: nasopharynx and nasal cavity, paranasal sinuses, middle ear and mastoid, and infratemporal space and pterygopalatine fossa. Symptoms from a parameningeal primary tumor are protean and include cranial nerve palsy, facial pain and swelling, meningeal irritation, nasal voice, mouth breathing, trismus, and painless adenopathy.81 Parameningeal primary tumors may erode skull base bone, transgress the dura, and invade brain; however, subarachnoid space dissemination is an uncommon event at diagnosis. Orbital tumors present with unilateral proptosis, chemosis, impaired mobility, ptosis, and lid or conjunctival mass.82 The other head and neck lesions present as a superficial mass that may cause seventh nerve palsy if arising in the parotid region or a mass lesion that interferes with speech or swallowing.
The second most frequent primary site is genitourinary (23%) divided between bladder and prostate (11%) and other genitourinary sites (female genital tract and paratesticular) (12%).78 The typical presentation is of bladder distention or outlet obstruction. Sarcoma botryoids may protrude from the vagina. Paratesticular presentation is as a painless mass. Next in frequency are extremity lesions (17%), which present with an enlarging, usually painless mass.
Routes of Spread
RMS is usually locally invasive and therefore frequently unresectable. In IRS-II patients with nonmetastatic tumor at presentation, 64% had only a biopsy or subtotal resection, approximately 20% had microscopic residual disease, and 16% had complete resection.80 A retrospective review of IRS-I and IRS-II patients in clinical groups I and II provided data on lymphatic metastases in 592 cases. Nodal spread was not influenced by gender, age, or histologic features. It was influenced by site as follows: 41% prostate, 26% paratesticular, 12% extremity, 3% trunk, 7% head and neck, 0% orbit, and 14% all sites.83 The inclusion of group I patients in the denominator minimizes the estimate of nodal involvement.
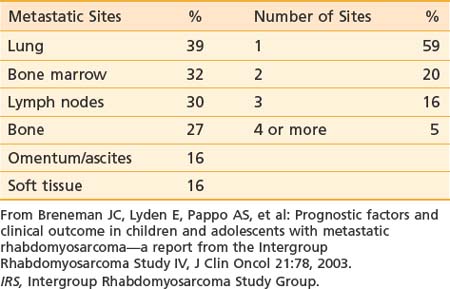
At presentation, 17% of IRS-II patients had hematogenous metastases (most common) or, less frequently, involvement of distant lymph node groups or malignant pleural or peritoneal effusion. Hematogenous metastases occur most frequently in lung. Both bone and bone marrow metastases are common.84 Unusual sites (e.g., heart, breast, and subcutaneous tissues) can occur.85,86 Brain metastases at diagnosis are rare.87 The pattern and frequency of metastatic sites for IRS-IV patients are shown in Table 55-2.
Diagnostic and Staging Studies
Patients with RMS require diagnostic studies that consist of both standard and site-specific assessments. In addition to the routine history, detailed family history of cancer should be obtained. In addition to the routine physical examination, an examination under anesthesia may be required for genitourinary presentations, and direct nasopharyngoscopy and laryngoscopy may be needed for upper airway primary tumors. Routine blood cell count and a standard chemistry assay are done. Renal function is assessed. Detailed assessment of the primary tumor with MRI or CT is required to determine location, invasiveness, and size. For orbit and parameningeal primary sites, the MRI should include the brain to characterize intracranial extension. Sagittal and coronal views are useful for radiotherapy planning.88 For genitourinary primary tumors, the entire urinary tract should be imaged with MRI or CT. Studies to assess dissemination include abdominal and chest CT and bone scan.89 A comparison of imaging with positron emission tomography to conventional imaging demonstrated that pet scanning was superior in detecting node and bone metastases.90 Bone marrow aspirate and biopsy are required, as is lumbar puncture for cerebrospinal fluid examination (chemistry and cytologic studies) for parameningeal sites.
Staging System
In the United States, 80% to 85% of patients with RMS are entered on the IRS trials that have been ongoing since 1972. The IRS grouping system for classifying patients in prognostic categories and for determining therapy is thus a de facto national standard. The IRS groups are described in Table 55-3. The system, which is surgical and pathologic, assesses extent of resection for patients with localized disease. It is clearly prognostic for treatment outcome but does not account for the variation in surgical aggressiveness as a first therapeutic event. Accordingly, the IRS committee designed a pretreatment staging system that separately categorizes the primary tumor (T), the regional nodal status (N), the presence of metastases (M), and the primary anatomic site. The system (Table 55-4) was retrospectively assessed and found to be of prognostic significance and to be reproducible.91,92 The TNM system was evaluated prospectively in IRS-III and was used to assign treatment in IRS-IV. The pretreatment staging system uses five features to determine stage: (1) primary site, (2) primary tumor invasiveness, (3) size, (4) nodal status, and (5) metastases. Primary sites are divided into favorable—orbit, nonparameningeal head and neck, biliary, and genitourinary (but not bladder or prostate); and unfavorable—all others, including bladder and prostate, extremity, and parameningeal. Invasiveness categories are T1, confined to anatomic site of origin; and T2, extension or fixation to surrounding tissues. T stage data are obtained, but they do not alter stage assignment. Size, rather than T stage, was believed to be a more reliable parameter and therefore affects stage assignment. Size (maximum tumor diameter) is divided into two categories: a, less than or equal to 5 cm; and b, greater than 5 cm. Nodal status is determined clinically: N0 indicates regional nodes clinically uninvolved, and N1 indicates regional nodes clinically involved. M staging assignment is M0, no distant metastases; and M1, indicating that metastases are present, often hematogenous, and including distant lymph nodes, pleural or peritoneal dissemination (or malignant effusion), and cytologically positive cerebrospinal fluid.
Table 55-3 Intergroup Rhabdomyosarcoma Study Clinical Group Classification
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|