Figure 10.6.1 Flow chart for methylation detection using bisulfite treatment and PCR. Bisulfite treatment converts unmethylated Cs to Us. Because PCR amplification then proceeds using a primer containing complementary As rather than Gs, the products of the bisulfite-treated unmethylated template will contain Ts in place of unmethylated Cs.
To accurately quantify methylated DNA at a specific locus, a real-time quantitative methylation specific PCR assay (Q-MSP; e.g., MethyLight, first described by Eads et al., 2000) can be used. In the Q-MSP assay, the amplification primers are designed to avoid hybridization to CpG sites, resulting in equivalent amplification of methylated or unmethylated DNA during PCR. Differentially dual-labeled fluorescent probes (i.e., Taqman probes) are used to distinguish between methylated and unmethylated DNAs. The accumulation of fluorescence during PCR amplification is proportional to the amount of methylated and unmethylated DNA in the original sample. The amounts of methylated and unmethylated DNA can be determined by interpolation of the crossing threshold for a particular sample on a standard titration curve run in parallel.
This unit provides procedures for treatment of genomic DNA with sodium bisulfite (Basic Protocol 1), conventional methylation-sensitive PCR (MSP; Basic Protocol 2), and real-time PCR (Q-MSP; Basic Protocol 3).
CAUTION: Radioactive, biological, and chemical substances require special handling; see APPENDIX 2A for guidelines.
BASIC PROTOCOL 1
SODIUM BISULFITE TREATMENT OF GENOMIC DNA
DNA is modified by sodium bisulfite treatment, converting unmethylated (but not methylated) cytosines to uracil. The basis for this reaction is that the sulfonation of position 6 in the cytosine ring results in the destabilization of the amine group at the 4th position, leading to deamination and conversion of the cytosine to uracil. Methylation of cytosine, which occurs at the fifth position of the cytosine ring, inhibits this reaction.
This reaction is absolutely dependent upon the denaturation of the DNA strands. Unmethylated cytosines in DNA that is not completely denatured will not efficiently be converted to uracil. After the purification of the DNA from the sodium bisulfite reaction, the DNA has to be desulfonated prior to PCR. This is simply done by treatment with NaOH followed by a second clean-up, e.g., ethanol precipitation.
Materials
Distilled water, room temperature and 65°C (autoclaved)
2 M and 3 M NaOH, freshly prepared (see recipe)
10 mM hydroquinone, freshly prepared (see recipe)
3.6 M sodium bisulfite, freshly prepared (see recipe)
Mineral oil
Wizard genomic DNA purification system (Promega), or equivalent, including:
0.5 M EDTA, pH 8.0
Nuclease-free water
Nuclei lysis solution
RNase A solution
Wizard SV lysis buffer
Wizard SV minicolumns
Wizard SV wash solution
10 mg/ml glycogen
3 M sodium acetate, pH 5.2
100% and 70% (v/v) ethanol, ice cold
54°C heating block
NOTE: All centrifugations, unless otherwise noted, are at maximum speed in a microcentrifuge (>20,000 × g).
NOTE: Before setting up the bisulfite reactions, it is important that all chemicals used in the reaction—sodium hydroxide, hydroquinone, and sodium bisulfite—be prepared fresh just before use. The amounts given can be scaled to produce the amount needed.
Prepare sodium bisulfite–modified DNA
Purify sulfonated DNA
Desulfonate DNA
6 µl of 3 M sodium acetate, pH 5.2
150 µl of 100% ethanol.
BASIC PROTOCOL 2
METHYLATION-SENSITIVE PCR
Following removal of sodium bisulfite and completion of the chemical conversion, the modified DNA is used as a template for PCR. The PCR used to assess DNA methylation can be carried out with a single primer pair, targeting either methylated or unmethylated DNA strands, or it can be multiplexed, with one primer pair targeting methylated DNA and the second primer pair targeting unmethylated DNA. In multiplex reactions, the primer pairs are designed to yield PCR products that are of discrete sizes. After PCR amplification, the bands are visualized by agarose gel electrophoresis and ethidium bromide staining. The presence of a band of the appropriate molecular weight indicates the presence in the original sample of either unmethylated alleles, methylated alleles, or both.
Any DNA sequence can be targeted for methylation analysis by appropriate design of PCR primers. The example given in Figure 10.6.2 is an assessment of DNA methylation at the imprinted SNRPN promoter. This assay is commonly used as a diagnostic assay for Prader-Willi syndrome and Angelman syndrome, and is a modification of a multiplex PCR procedure initially published by Kubota et al. (1997). A modification made to the initial method is that, in addition to the primers designed to hybridize to methylated and unmethylated SNRPN DNA after bisulfite treatment, a third primer pair specific for unmodified SNRPN DNA (SNRPNF and SNRPNR) has been included as an internal control to ensure that there is complete conversion during the sodium bisulfite reaction.
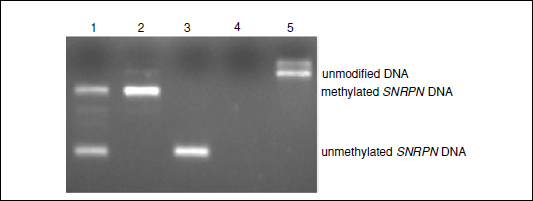
Figure 10.6.2 Analysis of DNA methylation at the imprinted SNRPN promoter. Lane 1, negative; lane 2, Prader-Willi syndrome; lane 3, Angelman syndrome; lane 4, water; lane 5, unmodified DNA.
NOTE: This procedure uses HotStar Taq DNA polymerase (Qiagen) and the 10× buffer and MgCl2 that come with the enzyme, for PCR amplification. Other Taq DNA polymerases and buffers from other manufacturers may also be used.
NOTE: As with all PCR methodology, exercise great care to prevent contamination of the preparations. Ideally, reserve a separate area for setting up PCR. See APPENDIX 2D for further discussion of special considerations for PCR experiments.
Materials
25 mM MgCl2
HotStar Taq DNA polymerase
PW/AS primer cocktail:
1 pmol/µl methylated MR1 DNA–specific primers (5′-AACCTTACCCGCTCCATCGCG-3′)
2 pmol/µl unmethylated DNA–specific PF2 primers (5′-GTAGGTTGGTGTGTATGTTTAGGT-3′)
2 pmol/µl unmethylated DNA–specific PR2 primers (5′-ACATCAAACATCTCCAACAACCA-3′)
1 pmol/µl unmodified DNA–specific SNRPNF primers (5′-GGAGGGAGCTGGGACCCC-3′)
1 pmol/µl unmodified DNA–specific SNRPNR primers (5′-GAAGCCACCGGCACAGCT-3′)
Sodium bisulfite–treated sample and control DNA (Basic Protocol 1)
3% (w/v) agarose gel
100- to 300-bp molecular size markers (e.g., Promega 100-bp ladder)
Ethidium bromide
UV transilluminator
2.4 µl of 25 mM MgCl2 (final 2 mM)
0.75 µl of 10 mM dNTPs (final 250 µM)
3 µl PW/AW primer cocktail (final 0.1 or 0.2 µM of each primer)
0.2 µl of 5 U/µl HotStar Taq DNA polymerase (final 1 U)
15.65 µl water.
Related posts:
 Variation in Humans
Variation in Humans
 locus in cancer and aging
locus in cancer and aging
 and dynamics of DNA methylation
and dynamics of DNA methylation
 Single-Molecule Mapping of Protein-DNA Interactions and DNA Methylation by MAPit
Single-Molecule Mapping of Protein-DNA Interactions and DNA Methylation by MAPit
 High-Throughput Arrays for Relative Methylation (CHARM)
High-Throughput Arrays for Relative Methylation (CHARM)
 Methylation Alterations in Multiple Myeloma as a Model for Epigenetic Changes in Cancer
Methylation Alterations in Multiple Myeloma as a Model for Epigenetic Changes in Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


