First author
Year
Anatomical site
HPV-
HPV+
Differently expressed genes (probes)
Analyzed genes (probes)
Oropharynx
Oral cavity
Tonsils
Base of tongue/tongue
Larynx
Hypopharynx
Keck
2014
X
X
X
75
55
1386
27,958
Mirghani
2014
X
15
15
224
(135,000)
Jung
2010
X
X
X
X
30
11 + 7
1498(2152)
>38,500
Lohavanichbutr
2009
X
X
78
41
(446)
>38,500
Martinez
2007
X
X
4
3
166
14,820
Pyeon
2007
X
X
26
16
92
>38,500
Schlecht
2007
X
X
X
X
30
12
149
(27,323)
Slebos
2006
X
X
X
X
28
8
91
>38,500
1.2.3 Regulation of Gene Expression by Epigenetics and MicroRNAs
Gene expression can be regulated at different levels. Here, we focus on differential epigenetic mechanisms in HPV+ve and HPV-ve HNSCC relevant to corresponding gene expression profiles. Epigenetic mechanisms are divided into three main groups: DNA methylation, histone modifications, and noncoding RNAs (ncRNAs).
As indicated by their name, ncRNAs are not translated into proteins. Highly abundant and functionally important RNAs belong to ncRNAs like tRNAs and ribosomal RNAs. Recently, many ncRNAs have been identified, but functional validations are often missing and some ncRNAs are considered to be non-functional (also referred to as Junk RNA). However, many ncRNAs are implicated in biological functions related to gene expression. Here, we focus on microRNAs, a group of trans-acting ncRNAs involved in regulation of gene expression.
MicroRNAs have been discovered in 1993 (Lee et al. 1993) and play a key role in posttranscriptional gene regulation in many cellular processes including cell division, development, cell death, and cell migration. They are transcribed by RNA polymerase II in the nucleus as primary microRNA (pri-miRNA) of 500–3,000 bases which are processed in a complex called Microprocessor by RNAse III (DROSHA) to generate 60–70 nucleotide precursor miRNAs (pre-miRNAs). This hairpin-like pre-miRNA contains the mature miRNA sequence in the double-stranded part of the stem loop. The pre-miRNAs are exported to the cytoplasm and further processed by DICER1 to produce the mature miRNAs, which are incorporated together with DICER1 and Argonaute (AGO) proteins in the miRNA-induced silencing complex (miRISC) (Fig. 1). Here, the miRNA directs the miRISC by sequence complementary to its target mRNAs and mediates gene suppression by targeted mRNA degradation and translational repression in so-called P‑bodies (processing bodies).
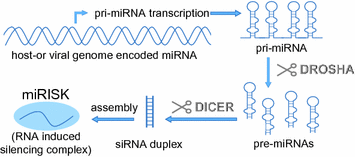
Fig. 1
Transcription and processing steps required for miRNA-induced silencing complex (miRISC) formation
Recently, it was shown that HPV genomes encode their own miRNAs (Gu et al. 2011; Qian et al. 2013a). The role of these miRNAs is still rather unclear, but target predictions mapped potential miRNA-binding sites to the HPV genome (within HPV genes E5, E1, L1 and in the LCR region) as well as to host target sequences and suggest multiple functions in cell cycle regulation, immune functions, cell adhesion/migration, and carcinogenesis (Qian et al. 2013a).
Altered miRNA expression has been implicated in various diseases including cancer, and overexpression of “oncogenic” miRNAs and downregulation of tumor suppressor miRNAs are related to carcinogenic processes like tumor formation, invasion, and metastasis. The biogenesis of miRNAs can be influenced at different steps during miRNA maturation. Pri-miRNA transcription can be positively or negatively influenced by genetic alterations, epigenetic modifications or tumor suppressors, and oncogenes. Further on, pri-miRNA processing, nucleolar export and final maturation steps can also be affected. Finally, miRNA function can be biased by competing endogenous RNA (ceRNA) or by mutations of miRNA-binding sites.
A PubMed literature search for “mirna & expression & cancer” results in more than 16.000 hits. However, this number drops to 26 by adding “head&neck&HPV” to the search term, and only few studies have analyzed differential miRNA signatures in HPV+ve compared to HPV-ve cancers. As in “classical molecular biology,” one cannot stick to the simplification that one miRNA has one target and therefore one function. Based on their rather small size, miRNAs may bind to several, often more than hundreds of more or less conserved target sequences. In addition, target genes can have numerous binding sites for different miRNA species, which makes gene regulation via miRNAs to a rather complex network of molecular interactions. Emerging techniques enlarge the knowledge on differential expressed miRNAs in HPV+ve compared to HPV-ve head and neck cancers; however, until now, data are rather inconsistent. Table 2 specifies miRNAs that were differentially expressed in HPV+ve compared to HPV-ve HNSCC in at least two publications. Given that one-third of the miRNAs are either upregulated in one study but downregulated in another study, shows that interpretation of miRNA data should be handled with care and experimental results highly depend on several factors like sample type, storage/processing, and analyzing techniques. Until now, no distinct picture of the role of miRNAs in HPV-associated HNSCC can be drawn, but their importance is obvious by considering the diverse and important functions of miRNA target genes in HNSCC (Table 2).
Table 2
Differential expressed miRNAs in HPV+ve compared to HPV-ve HNSCC
Reference | miRNA | Regulated in HPV+ compared to HPV- | Top-scoring targets (selected) based on number of validation methods (mirtarbase) | Target function |
|---|---|---|---|---|
hsa-miR-363 | Up | BCL2L11, CDKN1A, HI VEP1, CASP3, CD274 | Apoptosis, cell cycle, transcriptional regulation, immunology | |
hsa-miR-26b | Up and down | PTGS2, EPHA2, CCNE1, TAB1, RB1 | Prostaglandin biosynthesis, development, cell cycle, TGF-beta-interleukin 1- and WNT-1-signaling | |
hsa_miR_29a | Up and down | MCL1, DNMT3A, DNMT3B, BCL2, PIK3R1 | Apoptosis, DNA methylation, PI3K-signaling | |
hsa_miR_155 | Up and down | CEBPB, TAB2, TP53INP1, SMAD1, KRAS | Immune and inflammatory response, TGF-beta-TP53-signaling, cell growth, apoptosis, morphogenesis, development and immune responses, transformation | |
hsa-miR-222 | Up and down | CDKN1B, MMP1, KIT, PTEN, CDKN1C | Cell cycle, breakdown of extracellular matrix, proto-oncogene c-kit, tumor suppression | |
hsa-miR-125a | Down | ERBB3, CDKN1A, CD34, TP53, ERBB2 | Cell cycle, EGF signaling, cell attachment, tumor suppression | |
hsa-miR-143 | Down | KRAS, MAPK7, MYO6, DNMT3A, FNDC3B | Transformation, proliferation, differentiation, transcription regulation and development, intracellular vesicle and organelle transport, DNA methylation | |
hsa-miR-145 | Down | BNIP3, STAT1, FSCN1, KLF5, SOX2 | Apoptosis, cell viability, cell migration, motility, adhesion and cellular interactions, cell proliferation, embryonic development, cell fate, stem cell maintenance, epithelial-mesenchymal transition | |
hsa-miR-199a | Down | MET, MTOR, GSK3B, WNT2, HIF1A | Proto-oncogene, responses to DNA damage and nutrient deprivation, cell cycle arrest and immunosuppressive effects, transformation, energy metabolism, neuronal cell development, and body pattern formation, oncogenesis and development, cell fate, embryogenesis, hypoxia pathway | |
hsa-miR-126 | Down | VEGFA, SOX2, KRAS, PIK3R2, TERT | Proliferation and migration of vascular endothelial cells, embryonic development, cell fate, stem cell maintenance, epithelial-mesenchymal transition, transformation, PI3 K-signaling, telomere elongation | |
hsa_miR_181b | Down | TCL1A, TIMP3, PLAG1, BCL2, RNF2 | Development of mature T cell leukemia, inhibition of the matrix metalloproteinases, apoptosis, development and cell proliferation | |
hsa_miR_31 | Down | RHOA, SATB2, FOXP3, MMP16, HIF1AN | Tumor cell proliferation and metastasis, transcription regulation and chromatin remodeling, immunology, breakdown of extracellular matrix, oxygen sensing, HIF1A repression |
Besides ncRNAs, DNA methylation and histone modifications are two other important epigenetic processes affecting gene expression. In eukaryotes, methylation of cytosine typically occurs in a CpG dinucleotide and is associated with a number of key processes including genomic imprinting and X-chromosome inactivation, whereas methylation of adenine is restricted to prokaryotes. In normal development, gene expression is stably guided by DNA methylation during cell division and differentiation, which prevents differentiated cells to revert differentiation or to convert to another cell type. Gene expression can be affected by DNA methylation in two ways: Transcriptional proteins may be impeded in binding to a gene resulting in reduced gene expression. Second, methylated DNA may attract MBD (methyl-CpG-binding domain) proteins, thereby recruiting additional chromatin remodeling proteins like histone deacetylases. As a consequence, a compact, inactive chromatin structure is formed called heterochromatin, which links DNA methylation to histone modification, the third epigenetic processes affecting gene expression.
Histone proteins (2 copies each of the core histones H2A, H2B, H3, and H4) form a histone octamer, which is wrapped around by about 147 base pairs of DNA and forms a nucleosome core particle. Approximately 80 bases of DNA connect each nucleosome and linker histone proteins (e.g., H1) are involved in compaction of this chromatin structure. Histone proteins can be modified posttranslationally by acetylation, methylation, ubiquitination and phosphorylation of certain amino acids. These modifications affect molecular interactions between histones and between histones and DNA within the nucleosome core, which alters chromatin structure and thereby may affect gene expression in either an activating or inactivation manner.
DNA methylation and histone modifications in HNSCC and specific aspects for HPV-associated cancers are reviewed in other Chapters entitled “Risk factors for oral infection with Human Papilloma Virus” and “Predictive factors for outcome and quality of life in HPV-positive and HPV-negative HNSCC.”
2 Conclusion
Viral proteins interacting with key cellular regulators are important and necessary to drive HPV-associated carcinogenesis, contrasting with HPV-ve tumors where (simplified) each step in carcinogenesis has to be facilitated by genetic or epigenetic alterations. Consequently, mutations are less frequently found in HPV-associated cancers, but it is still not completely resolved, whether these are only passenger mutations or important at certain steps in carcinogenic progression.
HPV-driven cancers and HPV+ve OPSCC were shown to have recurrent focal 3q26.3-qter amplifications (Klussmann et al. 2009), which includes important cancer-associated genes such as TP63, SOX2, as well as the oncogene PIK3CA. In contrast, TP53 mutations, loss of chromosome arms 3p and 9p, and the amplification of 11q13 are prominently missing in HPV+ve tumors, while these changes are very common in HPV-ve tumors (Braakhuis et al. 2004). Importantly, the absence of chromosome 9p loss and the presence of HPV oncoproteins in HPV+ve tumors are requirements for overexpression of the p16INK4a gene, which serves as surrogate marker for HPV-associated cancers in the clinical setting. These molecular differences indicate different genetic progression models for both entities, but challenge the concept of field cancerization for HPV-related cancers.
Taken together, current molecular and clinical data clearly display HPV-related and HPV-unrelated HNSCC to be differential cancer subtypes. In addition, future research may provide evidence for additional subgroups, also within the HPV+ve HNSCC, which justify adapted therapy concepts for particular patient groups based on molecular (“omic-”) diagnostics in addition to tests performed by classical pathology.
References
Balz V, Scheckenbach K, Gotte K, Bockmuhl U, Petersen I, Bier H (2003) Is the p53 inactivation frequency in squamous cell carcinomas of the head and neck underestimated? Analysis of p53 exons 2-11 and human papillomavirus 16/18 E6 transcripts in 123 unselected tumor specimens. Cancer Res 63(6):1188–1191PubMed
Bayo P, Jou A, Stenzinger A, Shao C, Gross M, Jensen A, Grabe N, Mende CH, Rados PV, Debus J, Weichert W, Plinkert PK, Lichter P, Freier K, Hess J (2015) Loss of SOX2 expression induces cell motility via vimentin up-regulation and is an unfavorable risk factor for survival of head and neck squamous cell carcinoma. Mol Oncol 9(8):1704–1719. doi:10.1016/j.molonc.2015.05.006 CrossRefPubMed
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


