MATERNAL METABOLIC ADAPTATION TO PREGNANCY
Pregnancy is a period of significant maternal metabolic adaptations. Teleologically, the changes in maternal anatomy and physiology are thought to occur to support the growth and development of the fetus and prepare the mother for the physiological demands of pregnancy and lactation. The composite of changes are dynamic and evolve throughout the pregnancy.
Normal metabolic homeostasis
Metabolic fuels are derived from carbohydrates, fats, and proteins in the diet. All cells require a constant supply of fuel to provide energy for the production of ATP and cellular maintenance. After a meal, dietary components (glucose, free fatty acids, and amino acids) are delivered to tissues, taken up by cells, and oxidized to produce energy. Any dietary fuel that exceeds the immediate needs of the body is stored, mainly as triglycerides in adipose tissue, or glycogen in the liver, muscle and other cells, or to a lesser extent as protein in muscle. Between meals, substrates are drawn from stores and used as needed to provide energy. The regulation of body fuels is a complex interaction of nutrients and hormones that ensure a continuous supply of energy substrates with intermittent refueling or feeding.
Insulin and glucagon are the two major hormones that regulate fuel mobilization and storage. Insulin is a poly peptide synthesized as proinsulin in β-cells of the pancreatic islets and cleaved into insulin and c-peptide. Its primary role is to orchestrate the metabolism of not only glucose, but also of lipids and amino acids, which are vital for energy homeostasis. Insulin has anabolic and anticatabolic properties. In the liver, insulin promotes glycogen and fat synthesis, while suppressing glycogenolysis, and ketogenesis. In adipose tissue, it promotes fat storage and glycerol synthesis, and suppresses lipolysis. In muscle, insulin promotes glycolysis, glycogen and protein synthesis, and suppresses proteolysis. Glucagon, synthesized in the alpha cells of the pancreas, is a major counter-regulatory hormone of insulin. It is elevated when plasma glucose levels are low and promotes glucose production through glycogenolysis and gluconeogenesis. Epinephrine and cortisol are also insulin counter-regulatory hormones due to their anti-insulin effects.
Postabsorptive state
In the postabsorptive or fasting state, glucose-dependent tissues, like the brain, renal medulla, and certain formed blood cells, continually oxidize glucose as the primary fuel for energy. Because glucose is the preferred substrate for the brain, the maintenance of an adequate plasma glucose level is a physiologic priority. Low insulin levels result in a decrease in peripheral glucose uptake in tissues, such as adipose tissue and muscle. Initially, liver glycogen is degraded to provide glucose for glucose-dependent tissues. Approximately 70 g of glycogen is stored in the liver,1 while the total basal consumption of glucose is 200–250g/day,2 well in excess of stored hepatic glycogen. When the limited stores of glycogen are depleted, the liver uses carbon from lactate, glycerol, and amino acids to synthesize glucose through gluconeogenesis. Decreased insulin levels promote gluconeogenesis, and glucagon plays an additional role in the maintenance of continuous endogenous glucose supply. Glycogenolysis and gluconeogenesis increase to match the basal need for glucose for glucose-dependent tissues during fasting (Fig. 2.1 A).
Fig. 2.1 (A) In the fasting state, glucose for dependent tissues, like the brain and the fetus, is derived from the breakdown of hepatic glycogen stores. Once this reserve is depleted, glucose is produce de novo from amino acids released from protein stores in muscle. Free fatty acids (FFA) are released from adipose tissue, converted to ketone bodies in the liver and used to prevent excessive glycolysis in non-glucose dependent tissues. (B) Fed state. After the ingestion of a mixed meal, carbohydrates are broken down into glucose and other monosaccharides and taken up by all tissues. Any glucose that is not needed immediately for glycolysis is converted to glycogen or triacylglyerol and stored in liver, muscle, and adipose tissue for later use. Lipids are hydrolyzed to fatty acids, resynthesized to triacylglyerol (TG), and stored in adipose tissue. (C) Chronic over-feeding. Chronic over-nutrition and obesity can lead to adipocyte dysfunction and cellular inflammation. The release of various adipokines, including tumor necrosis factor (TNF)-α, results in insulin resistance in adipose tissue, skeletal muscle and liver. Insulin resistance in adipose tissue leads to lipolysis and increased FFA release, even in the presence of relatively increased insulin levels. With continued nutrient excess the capacity of adipocyte storage capacity is exceeded and lipid “overflows” to other tissues, such as muscle and liver, worsening insulin resistance and resulting in lipotoxicity and metabolic inflexibility.
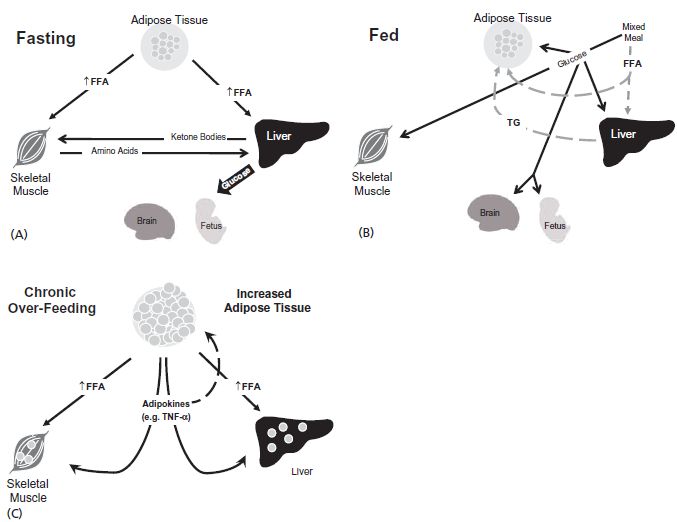
Insulin levels impact on the availability of all nutrients, including amino acids and fatty acids during periods of fasting. Low insulin levels allow for the increase in proteolysis and the augmentation of the release of amino acids from skeletal muscle, the primary reservoir of protein stores. The net flux of amino acids is from the muscle to the liver, with the gluconeogenic precursors, alanine and glutamine, accounting for the largest proportion of amino acids released.3 In adipose tissue, insulin inhibits hormone-sensitive lipase, which catalyzes the hydrolysis of stored triglycerides to free glycerol and free fatty acids. The consumption of free fatty acids in skeletal muscle is an important factor in limiting muscle glycolysis and glucose oxidation.
Post-absorptive state in pregnancy
Pregnant women have an added burden of supplying the growing fetus with energy substrates during periods of fasting. Glucose is the primary energy source for the fetus and the fetus is obligated to obtain most of the glucose it utilizes from maternal plasma due to the absence of significant gluconeogenesis.4 A carrier-mediated transport system (GLUT1)5 meets the high fetal demand with rapid transfer of glucose from the maternal compartment to the fetus. Maternal plasma glucose concentration and uterine/placental blood flow determine glucose supply to the fetus. Glucose transfer across the placental barrier is a relatively rapid process and has been defined as a flow-limited process.6
Fasting in pregnancy is more metabolically challenging for the mother due to the growing fetal demand for glucose as an energy substrate. During maternal fasting, plasma glucose levels decrease progressively with increasing gestational age.7 With short intervals of fasting, human pregnancy is marked by increased fasting plasma insulin levels and increased basal hepatic glucose production.8,9 A reduced insulin-induced suppression of hepatic glucose production may provide increased endogenous glucose production and therefore augments the supply of glucose for the mother and fetus between meals. Felig et al3 studied healthy women who were scheduled to undergo termination of pregnancy in the second trimester and healthy non-pregnant controls during a prolonged 84-hour fast. The fasted pregnant women had lower concentrations of plasma glucose and insulin, and greater ketone concentrations with prolonged fasting compared to the non-pregnant women. Felig’ s work in the 1970s led to the concept of “accelerated starvation” in pregnancy. The higher plasma ketones found in the fasted pregnant women were seen only in the presence of decreased insulin levels and presumably resulted from increased lipolysis.
Why are fasting glucose levels lower in pregnancy despite increased endogenous glucose production? The mechanism for this is not well understood. Decreased fasting glucose does not appear to be a result of decreased maternal protein catabolism based on urinary nitrogen excretion in pregnant compared to non-pregnant women.10 Maternal plasma alanine levels are decreased in fasted pregnant women compared to non-pregnant women and may represent the fetal siphoning of glucogenic precursors. Although protein catabolism is increased in pregnancy, increased utilization by the placenta and fetus is likely to cause a decrease in circulating glucogenic precursors.11 Some have suggested that the suppression of hepatic glucose production is not impaired in late pregnancy, but rather that the set point for plasma glucose levels is decreased.12
Postprandial state, non-pregnant
The changes in response to ingestion of a mixed macronutrient meal are based on homeostatic mechanisms that allow immediate usage or storage of fuel in expectation of periods of fasting (Fig. 2.1 B). Incretin peptides, such as glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1), are secreted from the gastrointestinal tract into the circulation in response to the ingestion of a meal, which enhances glucose-stimulated insulin secretion. Insulin release in the first phase acts predominately in the liver to decrease or shut down hepatic glucose production.13 Glucose uptake in the splanchnic bed is largely a result of increases in glucose availability, most of which will pass through the liver.14 Subsequently, increased insulin levels mediate peripheral glucose uptake, mainly in the muscle and adipose tissue.15 Larger amounts of insulin are required to affect peripheral glucose uptake than are needed to suppress hepatic glucose production.13 The repletion of muscle nitrogen depends on the net uptake of amino acids in muscle following a meal. In addition to its other functions, insulin acts to suppress proteolysis and accelerates the uptake of triglycerides, promoting fat synthesis and storage in adipose tissue and the liver. Postprandial increases in insulin levels promote the storage of all nutrients (glucose, amino acids, and fatty acids) for later use.
Postprandial state in pregnancy
In addition to the short-term (hour-to-hour) management of fuels, pregnant women have to regulate long-term energy balance that occurs with the changing metabolic demands of the mother and fetus throughout the pregnancy and during lactation. Early pregnancy is marked by storage of nutrients (anabolic state) in preparation for the later use of stored resources in the third trimester and during lactation when energy requirements increase (catabolic state). The energy balance adaptations in early to mid pregnancy probably result from large increases in estrogen, progesterone, and the lactogens (human placental lactogen and prolactin) (reviewed by Freemark16). The lactogens and progesterone increase appetite and induce hyperphagia, resulting in a 10–15% increase in food intake. Progesterone facilitates fat storage and the decline in pituitary growth hormone plays a permissive role in the deposition of body fat. The roles of lactogens and estrogen in lipogenesis are less clear and studies have been conflicting.16 Human placental lactogen stimulates hyperplasia and hypertrophy of beta islet cells. The resulting enhanced insulin secretion with normal peripheral and hepatic insulin sensitivity in early pregnancy promotes the storage of energy substrates through the inhibition of lipolysis, proteolysis, and glycogenolysis.
Overall, insulin sensitivity decreases progressively during pregnancy. Early and late pregnancy changes differ significantly. Although some debate exists about insulin action in early pregnancy, Catalano et al found no change in peripheral and hepatic insulin sensitivities in early pregnancy using the hyperinsulinemic – euglycemic clamp technique and glucose tracer, but glucose tolerance was improved.7,8,17 In early pregnancy, insulin secretion increases, while insulin action is variable and therefore, glucose tolerance may increase in some women.
Insulin resistance and a compensatory hyperinsulinemia are hallmarks of late pregnancy. Insulin-induced peripheral glucose uptake decreases 56% by the third trimester compared to the prepregnancy period, and insulin secretion increases 3 – 3.5 fold.7 Some animal18,19 and human studies20,21 have shown a reduction in insulin-induced suppression of hepatic glucose production in pregnancy, in contrast to others.12,22 Methodologic differences during insulin clamps are the likely explanation for the discrepancy, but the weight of evidence suggests that insulin’s ability to suppress hepatic glucose production is impaired in late pregnancy. Obese women with normal glucose tolerance have an impaired insulin-induced decrease in hepatic glucose production compared to lean counter par ts.21 In pregnant rodents, the accumulation of visceral fat contributes to the development of hepatic insulin resistance, an effect that may be modulated through the accumulation of hepatic triglycerides.23
INSULIN RESISTANCE IN PREGNANCY
The etiology of insulin resistance in pregnancy is not completely understood and is likely to be multifactorial. Historically, placental hormones have been implicated for many reasons. The extent of insulin resistance in pregnancy corresponds to the growth of the placenta and many placental hormones induce insulin resistance when given to non-pregnant individuals, including human placental lactogen (hPL),24,25 human placental growth hormone (hPGH),26 and progesterone.27,28 hPGH induces insulin resistance by antagonizing insulin action through regulation of p85 expression in adipose tissue.29 hPGH has been shown to cause severe insulin resistance in transgenic mice that express high levels of the hormone, similar to levels found in late human gestation.26 However, the transgenic mice in this study were also obese compared to wild-type animals, and the contribution of other factors, like visceral adiposity, have not been examined. Placental factors clearly have a role in the development of insulin resistance in pregnancy. Some hormones, such as hPGH, may directly affect insulin action; other factors may contribute indirectly to the insulin resistance through increased food intake and the promotion of lipogenesis.
The obesity epidemic and the increasing prevalence of the metabolic syndrome have spurred intense investigation into the adverse effects of increased adipose tissue and nutrient excess. An increase in adipose tissue, particularly an increase in visceral fat, is a key component of the metabolic syndrome30,31 and adipose tissue is no longer considered to be solely an energy storage depot. Normal pregnancy shares many common features with the metabolic syndrome, including increased adiposity, insulin resistance, hyperinsulinemia, and hyperlipidemia. Maternal body fat increases on average more than 3 kg32 over a relatively short time interval. Epidemiologic33,34 and animal23 studies suggest that visceral fat in particular increases in pregnancy, although descriptions of human body composition changes are limited due to increases in total body water and the restrictions of measurement modalities that can be used during pregnancy.35–38
Adipose tissue plays a role in regulating food intake, energy balance, and metabolic homeostasis through the production of fat-derived peptides. Several of these biologically active peptides affect energy homeostasis, such as leptin, which is expressed and secreted primarily by adipocytes. Leptin signals the adequacy of adipose stores to the hypothalamus, providing the afferent limb in energy homeostasis.39,40 The human placenta produces and secretes leptin into both maternal and fetal circulation,41 and the concentrations of leptin are elevated in pregnancy compared to the non-pregnant state, irrespective of body mass index,42 which may seem paradoxical because food intake is increased. “Lipostatic” signals, including leptin, become secondary and do not play a role in behavioral regulation of food intake if the energy supply (i.e. glucose) to the brain is threatened. As such, hyperphagia accompanies hypoglycemia independent of adipose mass.43 This phenomenon is termed “leptin resistance” and pregnancy is a “leptin-resistant” state. That the resistance to leptin in pregnancy is centrally mediated is supported by emerging evidence.44,45
Although adipocyte production of adipokines has a critical role in metabolic homeostasis, some adipokines may mediate the harmful biologic effects of increased adiposity. For example, TNF-α is associated with decreased insulin sensitivity in a number of conditions outside of pregnancy, including obesity46 and aging.47 In pregnancy, TNF-α plasma concentration is more predictive of insulin sensitivity than cortisol, human chorionic gonadotropin (hCG), estradiol, hPL, and prolactin.48 Althoug h 95% of TNF-α produced by the human placenta is released into the maternal compartment,48 TNF-α is secreted by both the placenta and adipose tissue, and the relative contribution of each source is uncertain.
Other adipokines (resistin, interleukin [IL]-1, and [IL]-6) have also been implicated as mediators of insulin resistance.49 Resistin, a newly discovered adipokine, decreases insulin sensitivity and increases plasma glucose levels.50,51 Higher plasma levels of resistin are found in women late in gestation compared to non-pregnant women, and term placentas have higher resistin mRNA expression compared with first trimester chorionic villi.52 However, the exact role of resistin and many other adipokines in pregnancy has yet to be determined.
Some adipokines improve insulin sensitivity (leptin, adiponectin. and peroxisome proliferator-activated receptor [PPAR]-γ). Little is known about the role of adiponectin during pregnancy. Although expression of adiponectin is specific to adipose tissue, plasma levels are lower in obesity.53 Serum adiponectin levels are lower in pregnant compared to non-pregnant women. However, no difference is found after correction for hemodilution.54 The roles of many adipokines and fat-derived peptides in pregnancy have yet to be elucidated.
NUTRIENT EXCESS AND METABOLIC DYSFUNCTION
The expansion of adipose tissue due to chronic over-nutrition and obesity can lead to adipocyte dysfunction, cellular inflammation, and insulin resistance (Fig. 2.1 C). In addition to the metabolic dysfunction caused by excess adipose tissue, the process of accumulating excess adipose tissue leads to metabolic dysregulation. Gregor and Hotamisligil55 have proposed that a pathologic excess of nutrients and excessive lipid storage in the adipocyte leads to loss of mitochondrial function and endoplasmic reticular stress. Adipocyte dysfunction results in insulin resistance, which may be mediated through the activation of c-jun N-terminal kinase (JNK) activation, inflammation, and oxidative stress. Further, when continued nutrient excess exceeds adipocyte storage capacity, lipid then “overflows” into other tissues.56 The oversupply of lipids into the liver, skeletal muscle and pancreatic islets results in a tissue-specific insulin resistance and impaired insulin secretion, generally termed “lipotoxicity”.56 In 1963, Randle et al57 proposed that increased fatty acid oxidation inhibits glucose oxidation and later, McGarry et al58 showed that hyperglycemia inhibits fatty acid oxidation. As a result of these two concepts, the concept of “metabolic in flexibility” has arisen, which proposes that in the setting of chronic over-nutrition, muscle tissue is unable to select the appropriate substrate for oxidation (glucose vs fatty acids) in response to the current nutrient supply,59 resulting in metabolic dysregulation in skeletal muscle, the primary tissue for peripheral glucose uptake in the non-pregnant state. This theory applied to pregnancy, a state of hyperphagia and rapid increases in maternal body fat, may have important implications.
INSULIN RESISTANCE AND GLUCOSE INTOLERANCE
The terms insulin resistance and glucose intolerance are often erroneously used interchangeably and should be differentiated. Insulin resistance refers to the reduced ability of insulin to act on target tissues. In the most basic terms, insulin is less effective in suppressing hepatic glucose production and greater amounts of insulin are needed to induce peripheral glucose uptake in the muscle and adipose tissue. In insulin-resistant states, more insulin is required to maintain glucose homeostasis.Glucose-intolerant states generally include some degree of insulin resistance and hyperinsulinemia, but the secretion of insulin is relatively inadequate for the degree of insulin resistance and the result is a mild fasting and/or transient postprandial elevations in plasma glucose levels.
In normal pregnancy, despite a well-demonstrated insulin resistance, the large compensatory increase in insulin secretion maintains maternal plasma glucose levels within a relatively narrow margin.20 Continuous glucose monitoring, which included more than 700 glucose measurements in a 72-hour period, demonstrated that normal weight, glucose-tolerant women at around 29 weeks of gestation had a mean fasting glucose level of 4.0 ± 0.7mmol/L (72.1 ± 13mg/dL) and a peak postparandial level of 5.9 ± 0.9mmol/L (106.2 ± 16mg/dL).60 Thus, although pregnancy is marked by insulin resistance, normal-weight women maintain serum glucose levels within a narrow margin. Women who are unable to compensate with increased insulin secretion become glucose intolerant. Further, glucose tolerance is a continuum. The detection of gestational diabetes is aimed at identifying pregnancies at risk for perinatal morbidity and to some extent to identify women at risk for Type 2 diabetes later in life. The threshold for maternal glycemia at which the risks for the fetus begin is unknown and is currently being debated (see chapters 6 and 7).
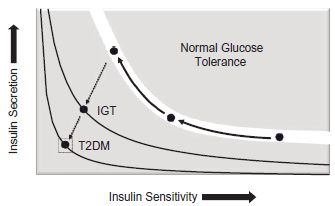
The relationship between insulin sensitivity and insulin secretion is reciprocal and non-linear in nature (Fig. 2.2). In order for glucose tolerance to remain unchanged, changes in insulin sensitivity must be matched by a proportionate yet opposite change in circulating insulin levels. With decreasing insulin sensitivity, as is seen in pregnancy, insulin secretion must increase for glucose tolerance to remain unchanged. Failure to secrete adequate amounts of insulin for the degree of insulin resistance results in a shift of the curve to the left and impaired glucose tolerance. This process underlies the development of diabetes.
Increasing insulin resistance and a compensatory hyperinsulinemia are progressive throughout the pregnancy.If insulin secretion cannot compensate for increased insulin resistance, glucose intolerance ensues. Many have used the analogy of the pick-up truck with a load in the bed of the truck. The engine of the truck symbolizes the pancreas and insulin secretion, and the load is insulin resistance. When the engine of the truck is normal (no defects in insulin secretion), the small load of insulin resistance from pregnancy itself is relatively inconsequential and does not affect the movement of the truck (glucose tolerance is normal). If other factors, like obesity, increased visceral fat or prolonged nutrient excess, contribute to a larger load, then the truck may be slowed, so that postprandial glucose levels are higher and may have a delayed return to baseline, allowing for increased transfer of glucose to the fetus. When the load is excessive, and the engine of the truck can no longer compensate or the engine is defective (beta cell dysfunction),overt diabetes results.
Fig. 2.2 To maintain normal glucose tolerance, insulin secretion must increase to compensate for decreasing insulin sensitivity during pregnancy (solid arrows). Failure to secrete adequate amounts of insulin for the degree of insulin resistance results in a shift of the curve to the left and impaired glucose tolerance (dotted arrows). This process underlies the development of diabetes (both gestational [IGT] and Type 2 [T2DM]). (Adapted from Kahn et al. Nature 2006; 444:840–6, with permission.)