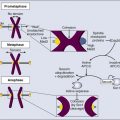
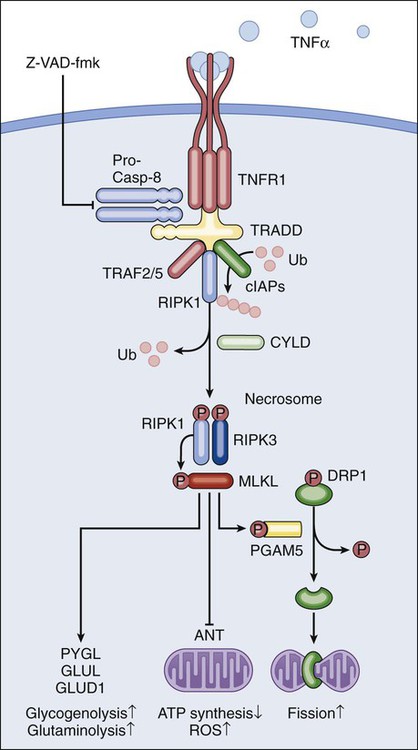
Lorenzo Galluzzi, Oliver Kepp and Guido Kroemer • Regulated cell death can occur via extrinsic apoptosis, intrinsic apoptosis, or regulated necrosis. Autophagy operates as a bona fide cell death mechanism in a few (mostly developmental) settings. • Oncogenesis results from multiple molecular alterations, one of which frequently impairs the ability of cancer cells to die in response to exogenous or endogenous signals. • Several oncoproteins and oncosuppressor proteins regulate the molecular machinery for apoptotic or necrotic cell death either directly or in an indirect fashion. • Targeting deregulated cell death signaling pathways in cancer is a clinical reality and underlies promising approaches for the development of novel anticancer regimens. Perhaps biased by their focus on the cell’s vital functions, biologists have disregarded the existence of programmed cell death (PCD; defined later) for a long time. Sporadic observations of PCD have been made throughout the nineteenth century by scientists such as Carl Vogt, August Weismann, Ludwig Stieda, Elie Metschnikoff, Walther Flemming, Sigmund Mayer, and John Beard.1 Nonetheless, the concept of PCD has been theorized as late as in the 1960s, thanks to the work of Sir Richard Lockshin.2 In 1972, John Kerr, Alastair Currie, and Andre Wyllie introduced the Greek term “apoptosis” (apo: from/off/without; ptosis: falling) to describe one type of cell death that manifests with peculiar morphologic traits.3 At that time and for the subsequent 30 years, apoptosis was thought to be the only form of PCD—an oversimplistic notion that recently has been invalidated.4, 5 In the mid-2000s, it became clear that other subroutines of cell death, notably necrosis, can occur in a regulated fashion and account for some instances of PCD.6 Regulated cell death constitutes a conserved mechanism whose utility trespasses evolutionistic barriers. For instance, in unicellular organisms that grow in colonies (such as yeast), the controlled demise of old and damaged cells increases the probability that fit individuals will survive adverse environmental conditions and hence will perpetuate their genes.7 Conversely, in metazoan (including humans), PCD is critical for embryonic and postembryonic development, as well as for the maintenance of adult tissue homeostasis.8 In line with this notion, defects in the molecular mechanisms that mediate cell death contribute to the development of a wide array of human diseases. On one hand, the excessive demise of postmitotic cells decisively contributes to the pathogenesis of diseases encompassing ischemia (of the heart and brain) and neurodegeneration. On the other hand, insufficient rates of cell death have been associated with autoimmune disorders and cancer.9 The first hint that the molecular machinery for cell death is involved in oncogenesis came in the mid 1980s, when it was found that a translocation between chromosomes 14 and 18 (t14;18) that is common in persons with lymphoma leads to the overexpression of the protein BCL-2.10 In subsequent work it was clarified that BCL-2 promotes lymphomagenesis by inhibiting the programmed demise of excessive B cells rather than by stimulating their proliferation.11 This work was the first demonstration that disabled cell death constitutes one of the hallmarks of cancer, irrespective of the histologic origin of malignant cells.12 During the subsequent two decades, it rapidly became clear that defects in the signaling pathways that lead to cell death not only contribute to oncogenesis and tumor progression but also determine, at least in some instances, the resistance of neoplastic cells to chemotherapy and radiotherapy.13 In cell death research, the word “programmed” is used to highlight the implication of one particular instance of cell death in developmental programs (and hence its physiological relevance), whereas “regulated” is used to stress the notion that one particular instance of cell death can be inhibited by targeted pharmacologic or genetic interventions (and hence is mediated by precise molecular mechanisms).4 Thus all instances of PCD are by definition regulated, but not vice versa. Additional recommendations on how to define specific cell death subroutines based on morphologic5 or biochemical4 parameters have been formulated recently and will be followed throughout this chapter. According to currently accepted models, two main types of cell death occur: apoptosis and necrosis. Additional cell death subroutines with very specific biochemical traits have been described, but they often constitute particular cases of apoptosis and necrosis and have a limited pathophysiologic relevance.4 Macroautophagy (hereafter referred to as autophagy) also has been indicated as a potential mechanism of cell death, a notion that remains highly debated.14 For a long time, instances of cell death have been catalogued as apoptotic on the basis of purely morphologic manifestations, including cytoplasmic and nuclear shrinkage (pyknosis), nuclear breakdown (karyorrhexis), and plasma membrane blebbing.5 This morphologic definition, reflecting the original observations by Kerr, Wyllie, and Currie,3 is progressively being abandoned in favor of a biochemical one.4 Thus apoptosis can occur as a result of the activation of either of two distinct but not mutually exclusive signaling pathways. On one hand, extrinsic apoptosis depends on a peculiar class of cysteine proteases called caspases and is always initiated by an extracellular stimulus acting on plasma membrane receptors. On the other hand, intrinsic apoptosis invariably follows the permeabilization of mitochondrial membranes but does not always require caspases.4 The enzymatic activity of executioner caspases, encompassing caspase-3, -6 and -7, is responsible for multiple (but not all) of the classic morphologic and biochemical manifestations of apoptosis, including karyorrhexis and the internucleosomal degradation of DNA.15 Extrinsic apoptosis is frequently elicited by the ligand-induced activation of plasma membrane proteins of the death receptor family, such as CD95/FAS and the tumor necrosis factor α receptor 1.16 Alternatively, apoptotic signals can be transduced by the so-called dependence receptors (such as patched 1) when the extracellular concentration of trophic factors, such as netrin-1, falls below a critical threshold level. This means that—at odds with death receptors—dependence receptors deliver proapoptotic signals in the absence, rather than in the presence, of their ligands.17 Death receptors undergo spontaneous trimerization because of the so-called preligand assembly domain.18 Ligand binding stabilizes this configuration and hence allows for the recruitment of several proteins at the cytoplasmic tails of death receptors, including (but in some instances not limited to) pro-caspase-8, receptor-interacting protein kinase 1 (RIPK1, also known as RIP1), FAS-associated protein with a death domain, and cellular inhibitor of apoptosis proteins (cIAP1 and cIAP2, which are E3 ubiquitin ligases that also inhibit apoptosis as a result of their ability to interfere with caspase activation).19 This multiprotein platform is known as “death-inducing signaling complex” and stimulates the conversion of pro-caspase-8 into caspase-8, which proteolytically activates caspase-3 and hence initiates the degradative cascade that mediates the execution of extrinsic apoptosis.20 Of note, the components of the signaling pathways that emanate from specific death receptors exhibit some degree of variation, yet all converge (in apoptosis-permissive conditions, as described later) to the activation of caspase-8. The molecular mechanisms that link dependence receptors to caspase-3 activation are not precisely understood, yet they appear to involve caspase-9 and the adaptor proteins DRAL and TUCAN (Fig. 5-1).21 Intrinsic apoptosis (also known as mitochondrial apoptosis) can be activated by a plethora of stimuli, including intracellular damage (to virtually any of the subcellular compartments) and by the so-called oncogenic stress (described later). Cells are equipped with a heterogeneous set of intracellular sensors that respond to perturbations first by activating signaling pathways for the reestablishment of homeostasis and the repair of damage and then, if damage is irreparable, by igniting intrinsic apoptosis.22 The central step in this cascade is regulated by the integrity of mitochondrial structure and function. Indeed, if proapoptotic signals are predominant, mitochondrial membranes get permeabilized, resulting in the abrupt cessation of adenosine triphosphate (ATP) synthesis and other metabolic functions, as well as in the spillage of several proteins that normally are confined in the mitochondrial intermembrane space.22 These proteins include cytochrome c (a semisoluble component of the mitochondrial respiratory chain), apoptosis-inducing factor (which normally contributes to the stability and function of respiratory complex I), endonuclease G, Smac/DIABLO, and HTRA2.23–27 Once in the cytosol, cytochrome c—together with dATP and the adaptor protein APAF1—drives the assembly of the apoptosome, a molecular platform for the activation of caspase-9.27 Smac/DIABLO and HTRA2 also stimulate caspase activation, though indirectly, by sequestrating and/or degrading cytosolic caspase inhibitors of the IAP family.23,24 Conversely, both apoptosis-inducing factor and endonuclease G translocate to the nucleus and mediate large-scale DNA degradation independently of caspases.25 According to current models, mitochondrial outer membrane permeabilization (MOMP) can proceed via two non–mutually exclusive mechanisms. First, MOMP can be initiated at the mitochondrial outer membrane (OM) by the pore-forming activity of proapoptotic multidomain proteins of the BCL-2 family.28 These proteins, such as BAX and BAK, contain several BCL-2 homology (BH) domains and a transmembrane domain that allow for their constitutive or inducible insertion into the OM. MOMP execution by BAK and BAX is regulated by other members of the same protein family. In particular, antiapoptotic proteins such as BCL-2, BCL-XL, and MCL-1 inhibit MOMP by binding to BAK and BAX and hence by maintaining them in an inactive conformation.29 Conversely, the so-called BH3-only proteins (small members of the BCL-2 family that often contain only the BH3 domain) can promote the pore-forming activity of BAK and BAX by two different mechanisms. Thus BH3-only proteins can either stimulate the conformational activation of BAX and BAK in a direct fashion or competitively displace BAX, BAK, or other BH3-only proteins from inhibitory interactions with BCL-2, BCL-XL, and MCL-1. BH3-only proteins (e.g., BID, BAD, and PUMA) are regulated at the transcriptional level (for instance, the gene coding for PUMA can be transactivated by the stress-responsive transcription factor p53), as well as by rapid posttranslational modifications (e.g., phosphorylation and proteolytic processing), de facto constituting sensors of intracellular stress that directly impinge on the regulation of intrinsic apoptosis.30 Second, MOMP can be initiated by an abrupt increase in the permeability to ions and small solutes of the mitochondrial inner membrane (IM), a process known as mitochondrial permeability transition (MPT). Although the actual physiological relevance of MPT and the underlying molecular mechanisms remain a matter of debate, MPT has been ascribed to the activity of a multiprotein protein complex that is assembled at the juxtaposition sites between the OM and the IM, the so-called “permeability transition pore complex” (PTPC).31 The main components of the PTPC, including the voltage-dependent anion channel of the OM, the adenine nucleotide translocase of the IM, and cyclophilin D (a protein of the mitochondrial matrix), normally mediate physiological functions. For instance, adenine nucleotide translocase catalyzes the exchange of adenosine diphosphate and ATP between the cytosol and the mitochondrial matrix. However, in response to specific lethal triggers, including cytosolic Ca2+ overload and the overgeneration of reactive oxygen species, the PTPC has been proposed to assume a high-conductance conformation, leading to the osmotic swelling of the mitochondrial matrix and consequent MOMP.22 Thus far, mouse knockout experiments failed to attribute a critical role in the regulation of intrinsic apoptosis to specific components of the PTPC, perhaps because of the existence of multiple and (at least partially) redundant isoforms of these proteins.34–34 One notable exception is represented by cyclophilin D, the absence of which has been shown to limit pathological cell death in multiple circumstances, in vitro and in vivo.35,36 Of note, the signaling pathways leading to extrinsic and intrinsic apoptosis exhibit some degree of cross talk. Thus, whereas in some cells including lymphocytes (type I cells), the activation of death receptors leads to pro-caspase-3 processing and apoptosis without any mitochondrial involvement,37 in other cells such as hepatocytes (type II cells), caspase-8 not only activates caspase-3 but also mediates the proteolytic cleavage of BID, generating a MOMP-inducing fragment.38 Thus in type II cells, MOMP functions as an amplifier of apoptotic signaling by eliciting the caspase-9-mediated activation of caspase-3. This cross talk is pathophysiologically relevant in vivo, as demonstrated by the fact that the hepatocytes of Bid-/- mice are partially protected from FAS-induced apoptosis (Fig. 5-1).39 Classically, necrosis was defined as an instance of cell death lacking the peculiar morphologic manifestations of apoptosis and the accumulation of cytoplasmic vacuoles that characterize autophagic cells. Somehow, this definition was in line with the belief that necrosis would always proceed in an unregulated fashion and would only terminate accidental instances of cell death.5 In the late 1990s, the group of investigators led by Peter Vandenabeele demonstrated that the engagement of FAS does not always lead to cell death via extrinsic apoptosis.40,41 This observation instilled in some researchers the suspicion that, similar to apoptosis, necrosis also might be orchestrated by a refined molecular machinery and ignited an intense wave of research that has not yet come to an end.6 The best-characterized pathway of regulated necrosis, which is also known as necroptosis, is elicited by the ligation of death receptors in conditions in which caspase-8 is inhibited (either by pharmacologic or by genetic interventions). In this context, death-inducing signaling complex–bound RIPK1 does not get degraded by caspase-8 as it occurs during extrinsic apoptosis; rather, it recruits and functionally interacts with its homolog RIPK3, generating the so-called necrosome.44–44 Recently it has been shown that the mixed-lineage kinase like protein and the mitochondrial phosphatase PGAM5 may contribute to the execution of regulated necrosis downstream of RIPK1 and RIPK3.45,46 These results confirmed previous suspicions regarding the involvement of mitochondria in regulated necrosis, driven by the prominent role exerted by reactive oxygen species and by metabolic circuitries that (at least in part) localize to mitochondria, such as glutaminolysis.44,47 Stimuli other than death receptor ligands (e.g., alkylating DNA damage) reportedly trigger regulated necrosis.48 However, it remains unclear whether these instances of regulated necrosis also require RIPK1 and RIPK3 and proceed via PGAM5-dependent mitochondrial fragmentation (Fig. 5-2).46 Regulated necrosis occurs during mammalian development, in particular at the bone growth plate (i.e., the zone of the bone that controls its length), as well as during adult tissue homeostasis, for instance, in the lower regions of intestinal crypts.49,50 Moreover, RIPK1/RIPK3-dependent necroptosis has been involved in the pathophysiology of several diseases, including viral infection, neurodegeneration, ischemia, and others.51 Autophagy entails the engulfment of intracellular structures (including organelles, protein aggregates, and portions of cytoplasm) by double-membraned vacuoles called autophagosomes. Autophagosomes normally fuse with lysosomes, leading to the degradation of their content by lysosomal hydrolases. Baseline levels of autophagy contribute to the maintenance of intracellular homeostasis by ensuring the removal of old and damaged (and hence potentially dangerous) organelles, notably mitochondria.52,53 In addition, autophagy is upregulated in response to a wide array of stressful conditions, including nutrient deprivation, hypoxia, and the presence of xenobiotics, such as anticancer agents.54 For a while it was thought that the continuative activation of autophagy eventually would lead to the exhaustion of cellular resources and cell death. Such an “autophagic cell death” was defined morphologically by an extensive vacuolization of the cytoplasm, representative of an elevated number of autophagosomes and autolysosomes (the organelles that are generated by the autophagosomal-lysosomal fusion).5,14 However, the association between the accumulation of autophagosomes and cell death has rarely if ever been proved to be causal, in particular in settings of stress-induced cancer cell death. Indeed, the inhibition of autophagy by pharmacologic or genetic means often accelerates (rather than inhibits) cell death, suggesting that autophagy constitutes a stress response mechanism that attempts (but fails) to avoid the cellular demise.55 These results cast doubts on the appropriateness of the term “autophagic cell death,” which inadvertently suggests a cause-effect relationship between these two phenomena.14 Thus far autophagy has been demonstrated to mediate cell death in several developmental scenarios, notably during the metamorphosis of insects.56,57 Moreover, at least in some instances, autophagy appears to contribute to the execution of human cancer cells succumbing to specific experimental cell death inducers in vitro.58 These observations de facto justify the use of the expression “autophagic cell death” under selected circumstances.4 Defects of the autophagic machinery have been associated with a plethora of human pathophysiologic conditions, including accelerated aging, neurodegeneration, and cancer.59,60 However, this association appears to be more strictly related to the role that autophagy exerts in the regulation of intracellular homeostasis rather than as a bona fide cell death mechanism52 and hence will not be treated here in further detail. According to classic models, single molecular alterations are per se unable to fully transform normal cells into highly aggressive cancer cells. Rather, oncogenesis seems to proceed along with a progressive increase in genetic instability and with the accumulation of several molecular defects. Often, if not always, one of these alterations consists of the interruption of the signaling cascades that ensure the homeostatic death of continuously proliferating cells.61 As they evolve, premalignant and malignant cells are indeed subjected to elevated levels of stress, in part as a result of the overactivation of cell-intrinsic oncogenic signaling pathways (so-called oncogenic stress) and in part because of microenvironmental conditions, which often are characterized by hypoxia and nutrient shortage (especially in rapidly proliferating neoplasms).62 Thus in virtually all scenarios, carcinogenesis requires the (at least partial) suppression of cell death signaling pathways. This suppression can result from loss-of-function mutations in proteins that transduce lethal signals or execute cell death (as with many oncosuppressor proteins) or from gain-of-function alterations in molecules that normally deliver prosurvival signals (as with several oncoproteins). Defects in the molecular pathways that regulate cell death also are instrumental to tumors when it comes to resistance to chemotherapy and radiotherapy.13 Oncogenes (i.e., genes that stimulate malignant transformation) were originally identified in tumorigenic viruses and then were shown to exist as inactive variants (or proto-oncogenes) in the human genome.63 Proto-oncogenes including MYC and NRAS are involved in the regulation of mitogenic signals and hence play a critical role in the control of tissue homeostasis. Proto-oncogenes are not intrinsically tumorigenic and must acquire gain-of-function alterations to become so. This process can originate from alterations as gross as chromosomal translocations that bring proto-oncogene coding sequences in the proximity of strong transcriptional regulators (as in the case of MYC, which is often rearranged in persons with lymphoma) or as specific as point mutations that render proto-oncogene products constitutively active (as in the case of NRAS, which is affected by hyperactivating mutations in 20% to 25% of all cancers).64,65 By transducing strong and persistent mitogenic signals, constitutively active MYC and NRAS—as well as other oncoproteins such as the epidermal-growth factor receptor (which is often overactivated in lung and colon cancer) and the Abelson tyrosine kinase (ABL, which is fused to BCR
Pathophysiology of Cancer Cell Death
Introduction
Fundamental Science—Mechanisms of Cell Death
Apoptosis
Extrinsic Apoptosis
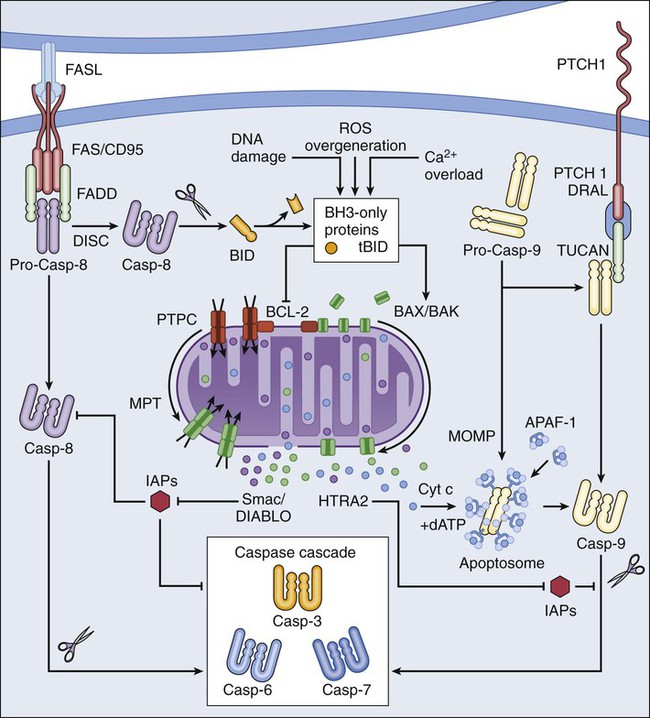
Intrinsic Apoptosis
Necrosis
Autophagy
Fundamental Science—Cell Death and Cancer
Oncogenes and Cell Death Regulation
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pathophysiology of Cancer Cell Death