Low grade
A. Malignant lymphoma, small lymphocytic
Consistent with chronic lymphocytic leukemia
Plasmacytoid
B. Malignant lymphoma, follicular, predominantly small cleaved cell
Diffuse areas
Sclerosis
C. Malignant lymphoma, follicular, mixed small cleaved and large cell
Diffuse areas
Sclerosis
Intermediate grade
D. Malignant lymphoma, follicular, predominantly large cell
Diffuse areas
Sclerosis
E. Malignant lymphoma, diffuse, small cleaved cell
Sclerosis
F. Malignant lymphoma, diffuse mixed small and large cell
Sclerosis
Epithelioid cell component
G. Malignant lymphoma, diffuse, large cell
Cleaved
Noncleaved
High grade
H. Malignant lymphoma, large cell immunoblastic
Plasmacytoid
Clear cell
Polymorphous
Epithelioid cell
I. Malignant lymphoma, lymphoblastic
Convoluted cell
Nonconvoluted cell
J. Malignant lymphoma, small noncleaved
Burkitt’s
Non-Burkitt’s
K. Miscellaneous
Composite
Mycosis fungoides
Histiocytic
Extramedullary plasmacytoma
Unclassifiable
Table 42.2
Kiel classification of non-Hodgkin’s lymphomas
B | T |
|---|---|
Low-grade malignant lymphomas | |
Lymphocytic | Lymphocytic |
Chronic lymphocytic leukemia | Chronic lymphocytic leukemia |
Prolymphocytic leukemia | Prolymphocytic leukemia |
Hairy cell leukemia | Small cell, cerebriform |
Mycosis fungoides/Sezary syndrome | |
Lymphoplasmacytic/cytoid (immunocytoma) | Lymphoepithelioid (Lennert’s lymphoma) |
Plasmacytic | Angioimmunoblastic (AILD) |
Centroblastic-centrocytic (follicular ± diffuse; diffuse) | T-zone lymphoma |
Centrocytic (mantle cell) | Pleomorphic, small cell (HTLV-1±) |
Monocytoid, including marginal zone cell | |
High-grade malignant lymphoma | |
Centroblastic | Pleomorphic, medium-sized and large cell (HTLV-I±) |
Immunoblastic | Immunoblastic (HTLV-I±) |
Burkitt lymphoma | |
Large cell anaplastic | Large cell anaplastic (Ki-1 +) |
Lymphoblastic | Lymphoblastic |
Almost as soon as the Working Formulation was published the application of newly developed immunophenotypic and molecular methods to the study of hematolymphoid neoplasms made it clear that the Working Formulation (and to a lesser extent the Kiel classification) was inadequate. The results of these studies showed that the categories of the Working Formulation were immunologically and molecularly heterogeneous. Almost immediately pathologists began to informally modify the Working Formulation by including immunophenotypic and molecular data. For example, the Working Formulation category of “malignant lymphoma, small lymphocytic” quickly had CD5+ and CD5− subsets. Other categories, such as “malignant lymphoma, diffuse mixed small and large cell” had B-cell and T-cell subsets. By the early 1990s, virtually all hematopathologists who used the Working Formulation were using a much improved, more complex version that included histologic, immunophenotypic, molecular, and cytogenetic data, but in a nonstandardized fashion. From here, a group of pathologists known as the International Lymphoma Study Group proposed a new lymphoma classification that was essentially a summary of the available literature and was built upon many of the concepts of the Kiel classification. This classification, first published in the journal Blood in 1994, was known as the Revised European-American Classification of Lymphoid Neoplasms (REAL) [12]. After a very important validation study of this classification organized by Dennis Weisenburger and James Armitage at the University of Nebraska [13], the REAL classification became the basis for a revised classification of lymphoid neoplasms by the World Health Organization (WHO), with the third edition in 2001 and the current fourth edition in 2008 (Tables 42.3, 42.4, and 42.5) [14]. There is a general consensus among the hematopathology and oncology community to use the WHO classification, although there are some areas that remain controversial. One aspect of the WHO classification, unconventional at the time of writing of the third edition in 2001, was that traditional NHLs were grouped with lymphoid leukemias, plasma cell myeloma, and HL, based on the common lymphoid origin of these neoplasms. In this chapter, we emphasize the pathology of traditional NHLs and HL that present as “lymphoma” as they are designated in the WHO classification, as the pathology of neoplasms that present as leukemia, plasma cell neoplasms, and histiocytic processes are covered in other chapters.
Table 42.3
2008 WHO classification of B-lymphoid neoplasms
Precursor B-cell neoplasms |
B lymphoblastic leukemia/lymphoma, not otherwise specified |
B lymphoblastic leukemia/lymphoma, with recurrent genetic abnormalities |
B lymphoblastic leukemia/lymphoma with t(9;22)(q34;q11,2); BCR-ABL1 |
B lymphoblastic leukemia/lymphoma with t(v;11q23); MLL rearranged |
B lymphoblastic leukemia/lymphoma with t(12;21)(p13;q22); TEL-AML1 (ETV6-RUNX1) |
B lymphoblastic leukemia/lymphoma with hyperdiploidy |
B lymphoblastic leukemia/lymphoma with hypodiploidy (hypodiploid ALL) |
B lymphoblastic leukemia/lymphoma with t(5;14)(q31;q32); IL3-IGH |
B lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3); E2A-PBX (TCF3-PBX1) |
Mature B-cell neoplasms |
Chronic lymphocytic leukemia/small lymphocytic lymphoma |
B-cell prolymphocytic leukemia |
Splenic marginal zone B-cell lymphoma |
Hairy cell leukemia |
Splenic B-cell lymphoma/leukemia unclassifiable |
Splenic diffuse red pulp small B-cell lymphoma |
Hairy cell leukemia-variant |
Lymphoplasmacytic lymphoma |
Waldenstrom macroglobulinemia |
Heavy chain diseases |
Alpha heavy chain disease |
Gamma heavy chain disease |
Mu heavy chain disease |
Plasma cell myeloma |
Solitary plasmacytoma of bone |
Extraosseous plasmacytoma |
Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT) |
Nodal marginal zone B-cell lymphoma |
Pediatric nodal marginal zone lymphoma |
Follicular lymphoma |
Pediatric follicular lymphoma |
Primary cutaneous follicle center cell lymphoma |
Mantle cell lymphoma |
Diffuse large B-cell lymphoma (DLBCL), not otherwise specified |
T-cell/histiocyte rich large B-cell lymphoma |
Primary DLBCL of the central nervous system |
Primary cutaneous DLBCL, leg type |
EBV + DLBCL of the elderly |
DLBCL associated with chronic inflammation |
Lymphomatoid granulomatosis |
Primary mediastinal (thymic) large B-cell lymphoma |
Intravascular large B-cell lymphoma |
ALK positive large B-cell lymphoma |
Plasmablastic lymphoma |
Large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease |
Primary effusion lymphoma |
Burkitt lymphoma |
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma |
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classical Hodgkin’s lymphoma |
Table 42.4
2008 WHO classification of T- and natural killer-cell neoplasms
Precursor T-cell neoplasm |
T-lymphoblastic lymphoma/leukemia |
Mature T-cell and NK-cell neoplasms |
T-cell prolymphocytic leukemia |
T-cell large granular lymphocytic leukemia |
Chronic lymphoproliferative disorder of NK-cells |
Aggressive NK cell leukemia |
Systemic EBV + T-cell lymphoproliferative diseases of childhood |
Hydroa vacciniforme-like lymphoma |
Adult T-cell lymphoma/leukemia |
Extranodal NK/T-cell lymphoma, nasal type |
Enteropathy-associated T-cell lymphoma |
Hepatosplenic T-cell lymphoma |
Subcutaneous panniculitis-like T-cell lymphoma |
Mycosis fungoides |
Sezary syndrome |
Primary cutaneous CD30+ T-cell lymphoproliferative disorders |
Primary cutaneous peripheral T-cell lymphomas, rare subtypes |
Primary cutaneous gamma-delta T-cell lymphoma |
Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma |
Primary cutaneous CD4+ small/medium T-cell lymphoma |
Peripheral T-cell lymphoma, not otherwise specified |
Angioimmunoblastic T-cell lymphoma |
Anaplastic large cell lymphoma, ALK+ |
Anaplastic large cell lymphoma, ALK− |
Table 42.5
2008 WHO classification of Hodgkin’s lymphoma
Nodular lymphocyte-predominant Hodgkin’s lymphoma |
Classical Hodgkin’s lymphoma |
Nodular sclerosis classical Hodgkin’s lymphoma |
Lymphocyte-rich classical Hodgkin’s lymphoma |
Mixed cellularity classical Hodgkin’s lymphoma |
Lymphocyte depletion classical Hodgkin’s lymphoma |
B-Cell Non-Hodgkin’s Lymphomas
B-Lymphoblastic Leukemia/Lymphoma, Not Otherwise Specified
B-lymphoblastic leukemia/lymphoma (LBL), not otherwise specified, is the most common type of B-LBL. Approximately 90 % of B-LBLs present as leukemic neoplasms, better known as acute lymphoblastic leukemia (ALL). By contrast, B-LBL without leukemic involvement at time of presentation is uncommon and represents no more than 10 % of all B-LBLs [15].
Patients with B-LBL are most commonly young children, under the age of 6 years, who present with ALL. Only a small subset of patients presents as lymphoma, with extranodal sites of involvement being most common, particularly skin, bone, and soft tissue [16–19]. Lymphadenopathy is an uncommon presentation of B-LBL and a mediastinal mass is rare. Regardless of stage, patients require aggressive chemotherapy regimens as are suitable for ALL [20].
Histologically, the normal architecture of the lymph node is effaced by a diffuse, relatively uniform proliferation of cells that have a tendency to stream out into perinodal tissues (Fig. 42.1) [16, 21]. The neoplastic cells have scant cytoplasm and fine nuclear chromatin. Tumor cell nuclei can be either convoluted or round. Mitotic figures are numerous. Due to the high growth rate, a starry-sky pattern secondary to individual cell necrosis and scavenging by macrophages can be present in 10–20 % of cases.
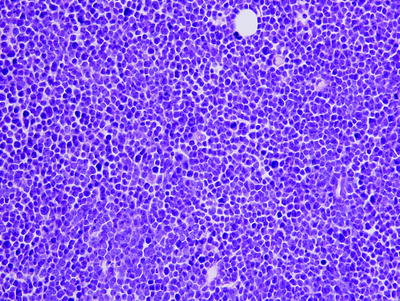
Fig. 42.1
B lymphoblastic leukemia/lymphoma involving adipose tissue. The neoplastic cells are small with blastic chromatin (hematoxylin–eosin, ×400)
B-LBLs are neoplasms with immature B-cell immunophenotypes that appear to correspond to “frozen” stages of normal B-cell maturation. Normal B-cells initially express the CD10 and CD19 antigens on the surface of the neoplastic cells, followed by CD22, and then CD20. These antigens are detectable in the cell cytoplasm prior to expression on the cell surface. The tumor cells usually do not express Igs on their cell surface [2]. These stages in B-LBL have been referred to as pro-pro B-cell, pro-B-cell, and pre-B-cell, with the latter expressing cytoplasmic IgM in addition to other B-cell antigens. Rarely, surface IgM or Ig light chain (usually not both) can be expressed. An extremely useful marker in the diagnosis of both B-cell (and T-cell) LBL is terminal deoxynucleotidyl transferase (TdT), a distinct type of DNA polymerase present normally only in immature lymphocytes [2, 22]. TdT has a physiologic role as it is involved in the process of gene rearrangement and is thought to add extra nucleotide bases between the variable (V), diversity (D), and joining (J) regions of the Ig and TCR genes undergoing rearrangement. TdT is expressed in almost all cases of B-cell (and T-cell) LBL.
Similar to surface antigen expression, the antigen receptor (Ig and TCR) genes appear to rearrange sequentially in normal B-cells (and T-cells) with a developmental hierarchy [23].At the earliest stage of B-cell differentiation the Ig heavy chain gene undergoes rearrangement. Subsequently, the Igκ light chain gene rearranges. If neither Igκ light chain gene allele is functionally rearranged, then the Ig λ light chain gene rearranges. This molecular mechanism allows a B-cell to express only one Ig light chain (the principle of allelic exclusion). The findings in B-LBLs mirror the normal state; all neoplasms carry monoclonal Ig heavy chain gene rearrangements, but only more mature tumors carry Ig light chain gene rearrangements [23]. Lineage infidelity is common in B-LBLs as T-cell receptor (TCR) gene rearrangements are common (TCR δ > TCR γ > TCR β) [24].
B-Lymphoblastic Leukemia/Lymphoma with Recurrent Genetic Abnormalities
A number of nonrandom cytogenetic and molecular findings occur in B-LBL cases, and some of these abnormalities are sufficiently common that the WHO classification now considers these as part of the definition of disease. In sum, there are seven types of B-LBL associated with specific molecular or cytogenetic abnormalities. Histologically, all of these neoplasms are indistinguishable from B-LBL not otherwise specified (NOS), and there are minor immunophenotypic correlations with specific abnormalities [25].
B-LBL with t(9;22)(q34;q11.2); BCR-ABL1
This neoplasm represents approximately 25 % of adult and <5 % of childhood B-LBLs [25, 26]. The prognosis of affected patients is relatively worse than that of patients with B-LBL NOS. Imatinib and other ABL1 tyrosine kinase inhibitors, in combination with other chemotherapy agents, are effective in patients with this neoplasm. Immunophenotypically, these neoplasms more commonly express the myeloid-associated antigens CD13 and CD33. CD25 is also commonly positive.
t(9;22)(q34;q11.2) joins the ABL1 gene at chromosome 9q34 with the BCR locus at 22q11.2, creating a BCR-ABL1 fusion gene that resides on the derivative chromosome 22 (also known as the Philadelphia chromosome). The result is a chimeric mRNA transcript and novel BCR-ABL1 protein with increased tyrosine kinase activity. Two types of t(9;22) occur. In approximately 25–50 % of adults with B-LBL, the translocation is identical to that seen in chronic myelogenous leukemia, and the fusion protein is 210 kd, known as p210. In the remaining 50–75 % of adults with B-LBL, the breakpoint on chromosome 22 does not occur in BCR but 100 kb upstream, resulting in a different fusion protein, p190, with greater tyrosine kinase activity and higher transforming capacity than p210.
B-LBL with t(v;11q23); MLL Rearranged
This type of B-LBL is the most common type in children <1 year or age and there is evidence that these translocations occur in utero in a subset of cases [25, 27]. The most common translocations involving 11q23 are t(4;11), t(9;11), and t(11;19). These translocations involve the HRX gene also known as ALL-1, MLL, or AF-4 on chromosome 11q23. Infants with translocations involving 11q23 present with a high white blood cell count, an increased frequency of central nervous system involvement, and a poor prognosis.
B-LBL with t(12;21)(p13;q22); TEL-AML1 (ETV6-RUNX1)
This type of B-LBL is common in children, representing 25 % of all B-LBL cases [25, 28]. These neoplasms are rare in infants and adults. Immunophenotypically, these neoplasms commonly express CD10, CD19, CD34, and the myeloid antigen CD13 and are usually negative for CD9 and CD20.
The t(12;21)(p13;q22) joins the ETV6 gene at chromosome 12p13 with the RUNX1 gene are 21q22. This event results in a novel ETV6-RUNX1 protein that is thought to act in a dominant negative fashion to impair differentiation. The t(12;21) most likely occurs very early in leukemogenesis because the translocation has been identified in neonatal blood spots of children who subsequently developed B-LBL.
B-LBL with Hyperdiploidy
This type of B-LBL is characterized by lymphoblasts that have >50 but <66 chromosomes, typically without other aberrations identified at the cytogenetic level [25, 29]. Most affected patients are children and the disease is rare in infants and adults. Prognosis is relative favorable. The immunophenotype is not distinctive. The presence of extra chromosomes present in these cases is not a random event: chromosomes 4, 14, 21, and X are common and chromosomes 1, 2, and 3 are rarely duplicated. The pathogenesis of this form of B-LBL is currently poorly understood.
B-LBL with Hypodiploidy (Hypodiploid ALL)
This neoplasm is characterized by lymphoblasts that have <45 chromosomes [25, 30]. This type of B-LBL is rare, representing <1 % of all B-LBL cases, and occurring in both children and adults. Within this category are cases of haploid B-LBL with 23–29 chromosomes. The immunophenotype is not distinctive. The prognosis of these patients is relatively unfavorable, with prognosis being the worst for patients with haploid B-LBL. The pathogenesis of this type of B-LBL is currently poorly understood.
B-LBL with t(5;14)(q31;q32); IL3-IGH
This type of B-LBL is rare, representing < 1 % of all B-LBL cases [25, 31]. The translocation juxtaposes the IL3 gene at chromosome 5q31 with the IGH gene resulting in overexpression of structurally normal IL3, thought to be responsible for eosinophilia that can be minimal or extensive, and in blood or bone marrow or both. Patients can be children or adults and prognosis is not thought to be different from patients with B-LBL NOS.
B-LBL with t(1;19)(q23;p13.3); E2A-PBX1 (TCF3-PBX1)
This type of B-LBL occurs in more commonly in children than adults and represents 6 % of all cases of B-LBL [25, 32]. In addition to B-cell markers and TdT, a subset of these cases has a pre-B-cell immunophenotype (cytoplasmic IgM+). Prognosis is similar to patients with B-LBL NOS.
This translocation involves the E2A (TCF3) gene on chromosome 1q23 and the PBX1 gene on chromosome 19p13.3. A novel chimeric gene resides on the derivative chromosome 19 and encodes a chimeric transcription factor and protein, E2A-PBX1. Rare cases in this category carry a t(17;19) involving the HLF gene.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Morphologically and immunophenotypically, the neoplastic cells of chronic lymphocytic leukemia (CLL) are identical to those of nodal-based small lymphocytic lymphoma (SLL), and the two entities are thought to represent different manifestations of the same disease [33]. Most patients who present with nodal-based SLL also have subclinical peripheral blood involvement, and progression to overt CLL frequently occurs. For these reasons, the WHO classification combined CLL and SLL together. Clinical data also suggest that patients with CLL or SLL can be treated similarly [34]. The term SLL is now applied rigorously to patients who have tissue involvement by a lymphoma that morphologically and immunophenotypically represents CLL/SLL, but with no evidence of leukemia.
CLL/SLL represents approximately 12 % of all B-cell lymphoid neoplasms [33]. CLL/SLL occurs primarily in middle-aged and older patients with a peak incidence in the sixth to eighth decades. The ratio of male to female cases is approximately 1.5:1. Clinically, CLL/SLL is associated with generalized lymphadenopathy. Although patients frequently have stage III or IV disease, CLL/SLL patients usually have an indolent clinical course with only vague symptoms such as weakness and anorexia; occasionally they have B type symptoms [10, 33, 34].
When involved by CLL/SLL, the normal lymph node architecture is usually totally effaced, but in some cases patent sinuses are present. At low power, vaguely nodular, pale areas are usually present (Fig. 42.2) that can be small or prominent, which represent proliferation centers (also known as pseudo-follicular growth centers or pseudofollicles) [35–38]. Cytologically, the neoplastic cells are predominantly small round lymphocytes with inconspicuous nucleoli, clumped chromatin, scanty cytoplasm, and infrequent mitoses. However, slightly larger lymphoid cells with irregular nuclear contours (known as prolymphocytes) and large cells with round vesicular nuclei and central nucleoli (known as paraimmunoblasts) are also found. These cells are most numerous in the proliferation centers. In some cases, these proliferation centers can surround residual benign germinal centers mimicking a marginal zone pattern (Fig. 42.1) [39].
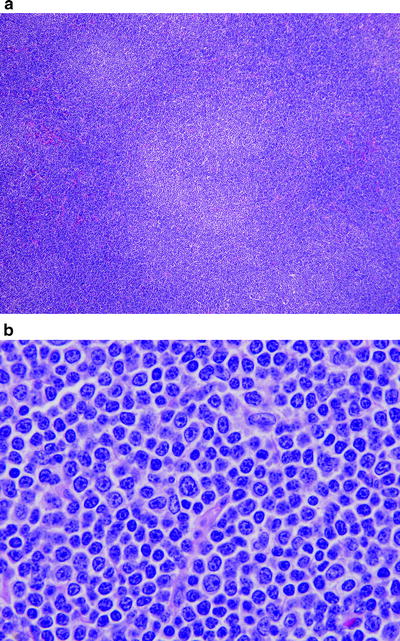
Fig. 42.2
Chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) involving lymph node. (a) Pale nodules seen at low magnification represent proliferation centers characteristic of CLL/SLL. (b) High magnification of proliferation center shows small lymphocytes, prolymphocytes (slightly irregular nuclear contours), and fewer large paraimmunoblasts (with nucleoli). (a) hematoxylin–eosin, ×100; (b) hematoxylin–eosin, ×400)
In tissue culture, CLL/SLL can be induced to secrete Ig, suggesting that these tumors are frozen in differentiation at a presecretory stage [40]. Furthermore, a small subset of CLL/SLLs exhibits plasmacytoid differentiation. Morphologically, the neoplastic cells in these cases have more abundant cytoplasm and the nucleus may be eccentrically situated. Like plasma cells, these cells contain cytoplasmic Ig that may be secreted resulting in a serum IgM paraprotein [41]. These patients typically have a low level of monoclonal paraprotein, less than 1.5 g/dl, and these neoplasms behave as do nonplasmacytoid cases of CLL/SLL.
Immunophenotypically, CLL/SLL are B-cell tumors that express monotypic Ig light chain (κ > λ), IgM, usually IgD, and pan-B-cell antigens such as CD19, CD20, CD22, CD79A, and PAX5 [2, 33, 42, 43]. The density of Ig and CD20 antigen expression on the surface of CLL/SLL cells is characteristically low corresponding to “dim” immunofluorescence detected by flow cytometry. BCL-2, CD21, and CD23 are usually positive, and CD79B and FMC7 are usually negative. These neoplasms also express the CD5 antigen, a pan-T-cell antigen that is not expressed on normal B-cells, but is expressed by CLL/SLL. Other T-cell antigens, CD10, BCL-6, and cyclin D1 are negative.
Genetic analysis of cases of CLL/SLL, with most of these patients presenting as CLL, has shown that this disease has a number of molecular subsets. The variable region genes of the Ig heavy and light chain genes may be mutated (>2 % change in germline sequence) or unmutated. Mutated cases of CLL are clinically indolent and patients have much longer median survival than patients with unmutated CLL [44, 45]. This may be attributable to defective B-cell receptor signaling in mutated CLL. As mutational analysis requires sequencing, a relatively labor-intensive procedure, surrogates of Ig mutational status have been sought. ZAP-70 and CD38 have been touted as reasonable surrogates, positive in unmutated cases of CLL. The correlation, however, is less than perfect [46].
A subset of cases of CLL/SLL, 5–10 %, appears to be familial although little is known about the genes involved or the type of inheritance. Chromosomal abnormalities have been shown in CLL/SLL and are best detected by fluorescence in situ hybridization (FISH) as CLL/SLL cells grow poorly in cell culture [47, 48]. Most FISH panels for CLL/SLL include probes to detect del(13q14), del(11q22) ATM, del (6q), del (17p13) P53, and trisomy 12. Del (13q14) is most common and is associated with a favorable prognosis. Deletions of 11q22/ATM and (17p13) P53 are associated with poorer prognosis. The pathogenetic genes involved in del(13q14), del(6q), and trisomy 12 are unknown. All together, approximately 75–80 % of CLL/SLL cases have abnormalities detected using FISH panels.
A small subset of CLL/SLL cases carries chromosomal translocations further contributing to the molecular heterogeneity of CLL/SLL. Overall, these cases represent less than 10 % of all CLL/SLL cases. Translocations involving the BCL-2 oncogene at 18q21 occur in <5 % of cases. More commonly, t(2;18) or t(18;22) translocations involving the Ig κ (chromosome 2p13) or the Ig λ (chromosome 22q11) light chain genes occur [49]. Rarely, t(14;18)(q32;q21) indistinguishable from that found in follicular lymphomas has been reported. The BCL-3 oncogene, involved in t(14;19)(q32;q13), has been identified in a small subset of CLL/SLL, and correlates with a poorer prognosis and the presence of trisomy 12 [50]. The t(2;11) involving BCL-11a occurs in 1–2 % of cases [51].
Approximately 5–10 % of CLL/SLL patients develop high-grade lymphoma, also known as Richter syndrome. Molecular findings in CLL/SLL cases that correlate with increased risk of progression to high-grade lymphoma include 8q24/MYC translocations and del(17p13)/P53 gene mutations [52, 53]. The most common histologic type of high-grade lymphoma in CLL/SLL patients is diffuse large B-cell lymphoma which is clonally related to the CLL/SLL in 50–60 % of cases [54]. Immune deficiency associated with CLL/SLL may be involved in patients who develop clonally unrelated diffuse large B-cell lymphoma. Other less common forms of Richter syndrome include the development of classical Hodgkin’s lymphoma, prolymphocytoid transformation, and rarely peripheral T-cell lymphoma [55, 56]. Specifically regarding prolymphocytoid transformation, approximately half of all cases once designated as B-cell prolymphocytic leukemia are currently better designated as prolymphocytoid transformation of CLL/SLL [33, 57].
Mu Heavy Chain Disease
In mu heavy chain disease, the neoplastic cells secrete a form of truncated IgM that cannot bind to Ig light chain, as a result of deletions in the VH or CH1 regions of the heavy chain [58]. Morphologically and immunophenotypically mu heavy chain disease resembles CLL/SLL, although the clinical presentation differs. Patients with mu heavy chain disease rarely have peripheral lymphadenopathy, and instead have a high frequency of hepatosplenomegaly. Bone marrow involvement also is present.
Lymphoplasmacytic Lymphoma
The terms lymphoplasmacytic lymphoma (LPL) and Waldenstrom macroglobulinemia have evolved in their usage over the past decade, with major changes from the 2001 to 2008 versions of the WHO classification. In the current version, LPL designates a lymphoma composed of small lymphocytes, plasmacytoid lymphocytes, and plasma cells that most often involves the bone marrow, but can involve lymph nodes, spleen, and uncommonly other extranodal sites [59]. A serum IgM paraprotein is also common in LPL. In the recent version of WHO classification, however, neither bone marrow involvement nor a serum IgM paraprotein is required for the diagnosis of LPL. Patients with LPL also can have an IgG or IgA serum paraprotein. Waldenstrom macroglobulinemia is considered to be a subset within the LPL category [59].
As the current definition of LPL is relatively new, clinical studies of LPL available in the literature focus primarily on patients with Waldenstrom macroglobulinemia. It also remains challenging to distinguish LPL from nodal marginal zone B-cell lymphoma, particularly in the absence of bone marrow involvement and a serum paraprotein. Lymph nodes involved by LPL usually retain their overall architecture. The pattern of LPL is diffuse and the cell population is predominantly small lymphocytes, with lesser plasmacytoid lymphocytes, and plasma cells. Intranuclear pseudoinclusions (Dutcher bodies) are common. Histologic evidence of other types of lymphoma that can be associated with a serum paraprotein, for example, proliferations centers as seen in CLL/SLL and monocytoid differentiation as seen in marginal zone B-cell lymphoma, must be absent.
The immunophenotype of LPL is B-cell, with the lymphocytic component expressing surface Ig and B-cell antigens and the plasmacytic component expressing cytoplasmic Ig and plasma cell-associated antigens including CD38 and CD138. The Ig heavy chain is usually IgM, but LPL cells can express IgG or IgA. BCL-2 is positive. The neoplastic cells are negative or usually negative for CD5, CD10, CD23, CD103, and BCL-6 [60].
Little genetic information has been reported for cases of LPL that do not meet the current definition of Waldenstrom macroglobulinemia. In most studies, cases of LPL and Waldenstrom macroglobulinemia have been lumped together (see text that follows).
Gamma Heavy Chain Disease
In gamma heavy chain disease, the neoplastic cells secrete a form of truncated IgG that cannot bind to Ig light chain, as a result of deletions in the VH or CH1 regions of the heavy chain [58]. Morphologically and immunophenotypically gamma heavy chain disease resembles LPL, although rare cases can be very plasmacytic and resemble plasma cell myeloma. Patients present with bone marrow involvement, lymphadenopathy, and/or hepatosplenomegaly similar to patients with LPL.
Waldenstrom Macroglobulinemia
Waldenstrom macroglobulinemia (WM) is specifically defined as LPL involving the bone marrow that is associated with a serum IgM paraprotein [59]. In other words, WM is a disease of the bone marrow in which a subset of patients has extramedullary disease. In the past, some investigators used WM to designate a clinical syndrome associated with a number of types of B-cell lymphoma that secrete serum IgM paraprotein [61]. This approach is no longer preferred and WM is now used in a specific fashion.
Clinically, patients with WM may present with a variety of symptoms and findings including mucous membrane bleeding, lymphadenopathy, hepatomegaly, peripheral neuropathy, and central nervous system abnormalities [59, 62–64]. Clinical and laboratory abnormalities that correlate with a poorer prognosis include: age ≥65 years, albumin <40 g/L, hemoglobin <11.5 g/dL, platelet count <100 × 109/L, beta-2-microglobulin >3 mg/L, and serum monoclonal protein concentration > 7.0 g/dL. These features have been combined into a prognostic index [65]. The serum monoclonal IgM paraprotein concentration is highly variable, with most patients having a paraprotein level greater than 1 g/dL. In our experience, approximately 5–10 % of patients present with signs and symptoms of a hyperviscosity syndrome [63]. Lymphadenopathy and hepatosplenomegaly occur in approximately 20 % of patients with WM. Leukemic involvement is usually absent, and when present the leukocyte count is usually normal. Lymphadenopathy in WM patients is generalized, but is usually modest.
Histologically, the bone marrow is usually replaced by WM in an interstitial or diffuse pattern, but nodular and rarely paratrabecular patterns of involvement occur. Lymph nodes involved by WM retain their general architecture. The neoplastic cells respect sinuses and tend to home to the medullary cord regions [59, 61, 66]. The capsule is extensively infiltrated, and perinodal adipose tissue is involved. Cytologically, three different types of WM have been described: lymphoplasmacytoid, lymphoplasmacytic, and polymorphous [67]. In the lymphoplasmacytoid type, the tumor cells are small with slightly increased cytoplasm characteristic of plasmacytoid differentiation. Cytoplasmic globules (Russell bodies) and intranuclear pseudoinclusions (Dutcher bodies) of IgM may be present. In the lymphoplasmacytic type, small lymphoid cells and mature (Marchalko-type) plasma cells are present. Russell and Dutcher bodies are common. These two types have no clinical significance. In the polymorphous type, large lymphoid cells are increased in the range of 5–10 %. Although the large cells do not form sheets and thus the criteria for large B-cell lymphoma are not present, patients with the polymorphous type have a poorer prognosis suggesting that this is an early stage of large cell transformation [67]. Amyloid deposition in the bone marrow or in extranodal sites occurs in small subset of WM patients.
Immunophenotypic studies have shown that WM cases have lymphocytic and plasma cell components [60, 68]. The lymphocytes are B-cells that express monotypic Ig light chain, IgM, pan-B-cell antigens, and BCL-2 and are negative for IgD, CD5, CD10, CD103, and BCL-6 [2, 33]. CD20 is often expressed variably (dim to moderate or partial). CD5 and CD23 can be dimly or partially expressed, best detected by flow cytometry, in approximately 50 % of cases [60, 68]. The plasma cell component expresses CD38 and CD138, and CD45/LCA is usually dim. In most cases, both CD19 and CD138 can be shown on the same cell population [60]. Immunohistochemical studies typically show a greater number of plasma cells than detected by flow cytometry. In a subset of cases of WM, approximately 25 %, plasma cells can predominate [60]. The lymphocyte and plasma cell components can be admixed or relatively segregated in the bone marrow biopsy specimen. After therapy, the plasma cells may persist with complete resolution of the lymphoid infiltrate [69].
The Ig genes are rearranged. No characteristic chromosomal abnormalities have been detected in WM. Numerical abnormalities have been reported; del(6q) is most common, in approximately 40–50 % of cases and trisomy 4 has been detected in approximately 20 %. Trisomies of chromosome 3 or 18 are rare [70–72]. Although the t(9;14)(p13;q32) was suggested as a marker of LPL/WM in the 2001 WHO classification, it is now clear that WM cases are consistently negative for the t(9;14)(p13;q32) as is now indicated in the 2008 WHO classification [59, 70, 71].
Follicular Lymphoma
Follicular lymphoma (FL) is the most common type of NHL in the United States [75]. The median patient age was 59 years in one study, with an age range from 23 to 90 years. Whites are affected more often than blacks. Unlike most other NHLs, women are affected equally or slightly more often than men [10, 75]. Most patients have clinical stage III or IV disease at the time of diagnosis. Involvement of the lymph nodes, spleen, and liver is common. Bone marrow involvement occurs in approximately one half of patients [10, 75]. A small subset of patients with FL develops clinical evidence of leukemia. The characteristic cell in the peripheral blood has a deeply clefted nucleus and has been referred to as a buttock cell [76]. Leukemic involvement only appears to influence prognosis when the leukocyte count is high. Subclinical involvement of peripheral blood by FL is detected commonly when assessed by molecular methods such as PCR. Rarely pediatric patients can develop FL. This patient subgroup has an excellent prognosis [77].
Histologically, the lymph node architecture is partially or completely effaced by neoplastic follicles, with a paucity of interfollicular tissue, and a large absolute number of follicles is the most reliable morphologic criterion for FL (Fig. 42.3) [78]. Unlike the lymphoid follicles in reactive hyperplasia, the follicles of FL are relatively uniform in size, lack a well-defined lymphoid cuff, and lack polarization [75, 78]. Histiocytes are usually less frequent in neoplastic follicles than in reactive follicles. Plasmacytoid differentiation can occur but is rare in FL [79].
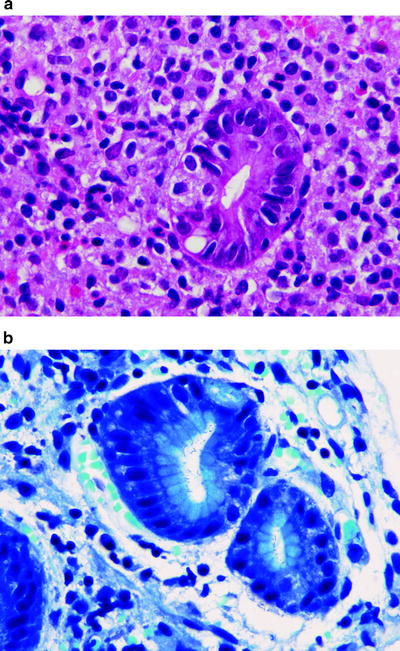
Fig. 42.3
MALT lymphoma involving the stomach. (a) Small lymphoma cells infiltrate a gland forming a lymphoepithelial lesion. (b) Giemsa stain highlights Helicobacter pylori species on surface of glandular epithelium. (a) hematoxylin–eosin, ×1,000. (b) giemsa, ×1,000)
Cytologically, FL cells closely mimic the cell types of normal germinal centers in reactive follicles [75]. Centrocytes have irregular, cleaved nuclear contours and coarse, condensed chromatin. Centrocytes can be small or large (also known as small or large cleaved cells). Centroblasts (also known as large noncleaved cells) are two to three times the diameter of centrocytes with vesicular nuclei and one to three nucleoli. The centroblasts are the proliferating component and therefore the number of mitotic figures correlates with the number of these cells. A small subset of FL, approximately 10 %, can exhibit marginal zone (also known as monocytoid) differentiation, manifested by a rim of monocytoid cells located peripheral to the neoplastic follicles. The presence of marginal zone differentiation has been shown to correlate with poorer prognosis [80].
Distinguishing between grades 1, 2, and 3 is based on a count of centroblasts as was originally described by Mann and Berard [81]. In grade 1, FL centroblasts are rare, <5/(high power) 400× microscopic field. Grade 2 tumors have ≥5 centroblasts/400× microscopic field and grade 3 tumors have >15/400× microscopic field. This system also has been modified by subdividing grade 3 FLs into two subsets, 3A and 3B [75, 82]. Grade 3B FL is composed of sheets of centroblasts without centrocytes in the background and appears to be closely related to de novo diffuse large B-cell lymphoma (DLBCL). The recent WHO classification has suggested that grade 1 and 2 cases of FL can be combined into so-called grade 1–2 as the clinical behavior of these two patient groups is similar [75]. Others suggest that grade 1, 2, and 3A are similar and can be considered as one group. These opinions will need to be confirmed with follow-up studies.
A component of diffuse pattern is common in biopsy specimens involved by FL, and in some cases the neoplasm can be entirely diffuse without a follicular component [75]. Thus, the presence of neoplastic follicles is highly characteristic of FL but is not required for diagnosis if the neoplastic cells otherwise have the cytologic, immunophenotypic, and molecular features of FL. Progression from a purely follicular to a follicular and diffuse pattern is common, and the presence of a diffuse component in patients with low-grade FL does not affect survival [83]. A diffuse component, however, usually accompanies an increased number of large cells and therefore a diffuse component is more common in patients with grade 3 FL, particularly grade 3B. When a patient with FL develops a grade 3 tumor that is entirely diffuse the neoplasm is designated as DLBCL. In biopsy specimens with both follicular and diffuse components it is useful to semiquantify the components, especially when the grade of the components is different, for example, follicular lymphoma, grade 1 (50 %) with DLBCL (50 %).
It is common for an individual patient with FL involving multiple sites to have discordant histologic findings. In this scenario, one biopsy site may be involved by low-grade FL and a second biopsy site may be involved by grade 3 FL or DLBCL. This was well shown in the days of performing staging laparotomy for patients with NHL [84]. Discordance is also common when comparing lymph node and bone marrow sites of disease. Lymph nodes may be involved by high-grade FL or DLBCL whereas the bone marrow is involved by grade 1 or 2 FL. In this scenario, patient survival is much better than if the bone marrow is involved by grade 3 FL, and therefore bone marrow disease should be graded when possible [85].
The term in situ follicular lymphoma describes a biopsy specimen involved by FL in which the follicles appear to be histologically benign, however, a subset of follicles are strongly BCL-2 positive and carry monoclonal IgH gene rearrangements supporting focal FL [86, 87]. Although the term is somewhat “catchy,” it is somewhat misleading as “in situ” in this context does not convey meaning similar to an in situ tumor at other anatomic sites (e.g., cervical carcinoma). Approximately one-third of patients with in situ FL have morphologically obvious FL at other sites, another third of patients develop FL subsequently, and one-third of patients have no other sites of disease at time of diagnosis of in situ FL or subsequently [87].
Immunophenotypic studies have shown that most FLs express monotypic surface Ig and Igκ is expressed more often than Igλ [2, 88]. Most tumors express IgM, but approximately 25 % express IgG or IgA, an expected finding since normal follicular center lymphocytes commonly undergo Ig heavy chain switching following exposure to antigen. IgD is usually negative, since its presence is a feature of naive B cells in the mantle zone. Typically, FLs express Ig and B-cell antigens at high density (bright immunofluorescence). A subset of FLs, mostly grade 3, can be negative for surface Ig.
FLs express pan-B-cell markers, and most are positive for the germinal center B-cell associated antigens CD10, BCL-6, germinal center B-cell expressed transcript-1 (GCET; also known as centerin), and human germinal center-associated lymphoma (HGAL), and are negative for T-cell antigens including CD5. Within the neoplastic follicles, meshworks of follicular dendritic cells can be appreciated that express CD21, CD23, or other follicular dendritic cell associated antigens. BCL-2 is expressed in 80–90 % of FL; as BCL-2 is negative in reactive germinal centers, this marker is helpful in differential diagnosis (Fig. 42.4) [89]. The centroblasts in FL commonly express activation and proliferation-associated (e.g., Ki-67) antigens. Koster and colleagues have suggested that proliferation rate assessed by Ki-67 immunostaining is prognostically more predictive than grading [90].
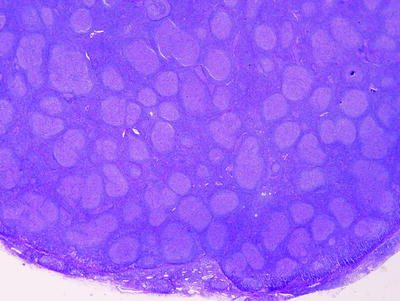
Fig. 42.4
Follicular lymphoma involving lymph node. The neoplasm forms follicles that are spread throughout the lymph node (hematoxylin–eosin, ×20)
The molecular hallmark of FLs is the t(14;18)(q32;q21) chromosomal translocation [4, 91]. Cytogenetic analysis has demonstrated t(14;18) in over 90 % of cases. In this abnormality, the bcl-2 oncogene on chromosome 18q21 is juxtaposed with the joining region of the IgH gene on chromosome 14q32. The bcl-2 gene is deregulated, by being placed under the influence of IgH gene regulatory elements (perhaps the enhancer region). The presence of t(14;18) alone does not appear sufficient for neoplastic transformation since this translocation has been identified in rare cells in the tonsils, lymph nodes, and blood of normal individuals without clinical evidence of lymphoma [92].
The BCL-2 protein is a 25-kd molecule that is overexpressed in FLs and protects cells from programmed cell death (apoptosis). The inhibition of apoptosis prolongs cell life, resulting in an expanded compartment of B-cells at increased risk for additional molecular defects, presumably involved in neoplastic transformation [89].
Fluorescence in situ hybridization (FISH) is an excellent way to demonstrate the t(14;18)(q32;q21) as the probes are large and cover most breakpoints [4]. There are at least three breakpoint clusters in the BCL-2 gene for which PCR assays for clinical laboratories have been developed. The two most commonly assessed breakpoint regions are the major (MBR) and minor (MCR) breakpoint cluster regions, involved in 50–60 % and 5–10 % of cases, respectively [4]. An intermediate cluster region (ICR) also is involved in 10–20 % of cases.
Gene expression profiling using cellular RNA and cDNA microarrays has been performed on cases of FL. The most important finding of these studies is that the host microenvironment, rather than the tumor cells themselves, has prognostic significance. Molecular signatures corresponding to host T-cell subsets and macrophages are associated with better or worse survival [93, 94]. Another important finding is that gene expression patterns are different in tumors with the t(14;18)(q32;q21) versus tumors without this translocation [95].
Extranodal FL
The comments related to the pathology of FL, to this point, have been confined to nodal disease. FL can also arise at extranodal sites. Although the morphologic features of FL at extranodal sites resemble neoplasms in lymph nodes, there are important immunophenotypic and molecular differences that suggest that extranodal FLs are somewhat different diseases [75]. Extranodal sites for FL of particular interest include the skin, small intestine, and testis. These tumors, when localized, are commonly BCL-2 negative and lack the t(14;18)(q32, q21). Skin FLs are now separately classified in the current WHO classification [14].
Extranodal Marginal Zone B-Cell Lymphoma of Mucosa-Associated Lymphoid Tissue (MALT Lymphoma)
Prior to the advent of immunologic and gene rearrangement techniques, the diagnosis of extranodal low-grade B-cell lymphoma was established infrequently. Extranodal infiltrates composed of small round or slightly irregular lymphoid cells, often admixed with plasma cells, histiocytes, and lymphoid follicles were often classified as pseudolymphomas, since clinical studies showed that patients with these lesions pursued an indolent clinical course. Immunophenotypic and gene rearrangement studies have since shown that approximately 60–70 % of pseudolymphomas express monotypic Ig light chain and contain Ig gene rearrangements [98, 99]. Thus, these tumors are now classified as low-grade B-cell lymphomas, and in most cases represent extranodal marginal zone B-cell lymphomas of MALT (MALT lymphoma).
MALT lymphoma was originally described by Isaacson and Wright [100] as a subset of gastrointestinal lymphomas in European patients that resembled immunoproliferative small intestinal disease (IPSID, also known as Mediterranean lymphoma). Similar neoplasms arising in the lung and salivary gland were then identified shortly thereafter suggesting that the common aspect of these tumors was that they arose from lymphoid tissue associated with mucosal surfaces [101]. Subsequently, MALT lymphomas were identified in a variety of extranodal sites including the thyroid gland, thymus, breast, conjuctiva, gallbladder, cervix, larynx, trachea, dura, skin and kidney, as well as other sites [102]. Thus, the term MALT lymphoma, although it continues to be used, is somewhat misleading in the sense that not all MALT lymphomas arise in sites involving mucosal surfaces. Given this wide number of sites involved, the diversity of pathogenesis, and the varied molecular findings (see text that follows), one can now make the argument that these neoplasms should no longer be grouped together as an “entity.” However, these tumors do share morphologic and immunophenotypic similarities and are indolent clinically. MALT lymphomas represent approximately 7–8 % of all NHL [102].
Patients with MALT lymphoma tend to have localized disease for prolonged intervals before disseminating [102]. This is particularly true for patients with gastric MALT lymphoma whereas patients with nongastric tumors more often present with stage III or IV disease or disseminate more often [103–105]. Anatomic sites that are commonly involved by nodal low-grade B-cell NHLs, such as the bone marrow, liver, spleen, and peripheral blood, are infrequently involved by MALT lymphomas. With prolonged follow-up, patients with MALT lymphoma relapse frequently, and relapses often occur in other extranodal sites [102, 106]. In a multivariate analysis at M.D. Anderson Cancer Center, factors that independently predicted overall survival included: elevated serum beta-2 microglobulin level, presence of B symptoms, and male gender [107].
Histologically, four findings are present in most MALT lymphomas: a population of neoplastic small lymphoid (centrocyte-like) cells, occasional large lymphoid cells, reactive lymphoid follicles, and lymphoepithelial lesions when the MALT lymphoma involves a site that normally has glandular epithelium (Fig. 42.5) [101, 102]. The neoplastic small lymphoid cells exhibit a range of cytologic appearances. In some neoplasms these cells closely resemble small lymphocytes, with or without plasmacytoid differentiation. In other neoplasms, the tumor appears biphasic: one component is small lymphoid cells with minimal cytoplasm, and the other component exhibits extensive plasmacytoid differentiation with many cells resembling mature plasma cells. Plasmacytoid differentiation tends to be more prominent in nongastric tumors. In other tumors, the neoplastic cells have markedly irregular nuclear contours and resemble small cleaved lymphocytes (centrocyte-like cells). All of these neoplastic cell types may have abundant pale cytoplasm with well-defined cytoplasmic membranes imparting a monocytoid appearance.
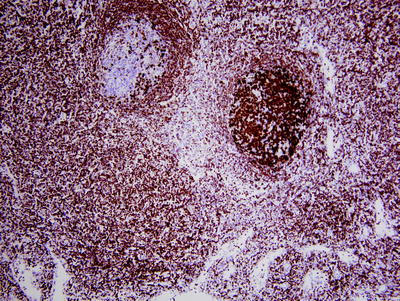
Fig. 42.5
Follicular lymphoma (FL) in situ involving lymph node. In routine histological sections, this lymph node looked normal. BCL-2 immunostaining of the two follicles in this field shows a reactive follicle on the left (BCL-2 negative) and in situ FL on the right (BCL-2 strongly positive). (immunohistochemistry with hematoxylin counterstain, ×100)
In most MALT-lymphomas, occasional large lymphoid cells are also present, which is not surprising since the natural history of many low-grade lymphomas is to accumulate large cells and then progress to a high-grade lymphoma. When the large cells are numerous and form confluent sheets, the neoplasm has evolved to diffuse large B-cell lymphoma. Use of the term high-grade MALT-lymphoma is not recommended in the WHO Classification [102].
Reactive lymphoid follicles are also usually present in MALT lymphomas, usually surrounded by neoplastic small lymphoid cells. These neoplastic cells may accumulate in the follicles (termed colonization), and the tumor acquires a nodular low-power appearance [108]. These neoplasms are not truly of follicle center cell origin. When MALT lymphomas involve a site that normally has epithelium (e.g., stomach), the neoplastic cells have a marked tendency to infiltrate epithelium, forming so-called lymphoepithelial lesions. In these lesions, three or more neoplastic B-cells are found within the epithelium, usually associated with evidence of epithelial damage. In addition to these common histologic findings in MALT lymphomas, there are also important site-specific differences.
In the stomach, normal lymphoid tissue is not present; however, benign MALT is acquired, often in response to Helicobacter pylori infection (Fig. 42.5) [102, 109, 110]. Approximately 30 % of cases of H. pylori-induced chronic gastritis are associated with lymphoid follicles; a smaller subset of cases also develops lymphoepithelial lesions. Conversely, over 75 % of gastric MALT lymphomas are associated with H. pylori infection. Thus, it seems likely that H. pylori induces the development of benign MALT and predisposes to the development of MALT lymphoma. Studies also have shown that benign MALT tissue and MALT lymphomas often regress following antibiotic therapy appropriate for H. pylori [111].
Other associations between infectious agents and MALT lymphoma have been identified. Chlamydia psittaci has been associated with up to one-third of MALT lymphomas in the ocular adnexal region, Borrelia burgdorferi with a subset of cutaneous MALT lymphomas, and Campylobacter jejuni in MALT lymphoma involving the small intestine [112–114]. There are other studies, however, showing a low frequency of infectious organisms in patients with MALT lymphoma. Possibly geographic differences explain these discrepancies.
In the normal adult lung, MALT tissue is also poorly developed and inflammatory conditions usually precede the development of MALT lymphoma. Two inflammatory diseases are frequently associated with lung MALT lymphoma, Sjogren syndrome and lymphoid interstitial pneumonia [102]. Similarly, MALT lymphomas of the salivary gland are usually associated with Sjogren syndrome and Hashimoto thyroiditis frequently precedes MALT lymphoma of the thyroid gland. Patients with Sjogren syndrome have a 44-fold increased risk of developing lymphoma, most of which are MALT lymphomas [102, 115]. Similarly, patients with Hashimoto thyroiditis have a 70-fold increased risk of lymphoma involving the thyroid gland, most of which are MALT lymphomas [102, 116].
In other organs, MALT lymphomas are less well understood. For example, in the kidney MALT lymphomas are known to occur, and share histologic and molecular features with MALT lymphomas at other sites, but there is no clear association with an underlying inflammatory condition or infectious organism [117]. In the breast, despite the presence of breast epithelium, it is unusual to identify lymphoepithelial lesions and MALT lymphoma-associated translocations are rare [118].
Immunophenotypic studies have shown that MALT lymphomas express monotypic Ig light chain, usually IgM, pan-B-cell antigens, and BCL-2. These tumors typically do not express IgD, CD10, BCL-6, cyclin D1, or T-cell antigens including CD5. There are no specific immunophenotypic markers for MALT lymphoma. BCL-10 is expressed by MALT lymphomas and the pattern of expression is stated to predict the presence of specific chromosomal translocations [119]. In our experience, however, although there is a correlation between translocations and the BCL-10 staining pattern, there is also a substantial false positive rate if only BCL-10 staining is used. This lack of specificity also may be true of certain anatomic sites of MALT lymphoma rather than for all sites of disease.
MALT lymphomas carry monoclonal Ig gene rearrangements. In a small study of four patients with multiple sites of disease, VDJ sequence analysis of the IgH gene showed that different sites of MALT lymphoma were not clonally related in 3 of patients [120]. Approximately 10 chromosomal translocations have been characterized or partially characterized in MALT lymphomas [121], and these translocations, in aggregate, have been shown in approximately 30–40 % of MALT lymphomas. These data indicate that MALT lymphoma is highly heterogeneous at the molecular level. Four translocations, to date, are relatively well characterized.
The t(11;18)(q21;q21) has been identified in 20–30 % of MALT lymphomas [122, 123]. In this translocation the API2 gene on 11q21 and the MALT1 gene on chromosome 18q21 are disrupted and recombined to form a novel API2-MALT1 fusion gene. The API2 gene belongs to a protein family known as inhibitors of apoptosis proteins (IAP) that are evolutionary conserved and play a role in regulating apoptosis. MALT1 is a novel gene of unknown function, but is critical to the function of API2-MALT1, which is known to activate NF-κB [124]. The t(11;18) is most common in MALT lymphomas of the stomach and lung [125].
The t(14;18)(q32;q21) occurs in 10–20 % of MALT lymphomas, most often involving the ocular adnexal region, skin, and salivary glands [125, 126]. This translocation juxtaposes MALT1 at chromosome 18q21 with the IgH gene on chromosome 14. This translocation has been implicated in NF-κB activation [124]. The t(3;14)(p14.1;q32) has been reported in up to 10 % of MALT lymphomas and most commonly is found in tumors involving the ocular adnexal region, thyroid gland, and skin [127]. t(1;14)(p22;q32) is an uncommon translocation identified in less than 5 % of MALT lymphomas that juxtaposes on intact BCL-10 gene at 1p22 adjacent to the IgH gene at 14q32 [128]. The translocation truncates BCL-10 and thus BCL-10 protein loses its pro-apoptosis function. The t(1;14) occurs most often in MALT lymphomas of the small intestine. BCL-10 gene mutations also occur outside the context of the t(1;14) in 7–10 % of MALT lymphomas [129]. These mutations consist predominantly of deletions or insertions and are predicted to result in truncated proteins.
MALT lymphomas can be divided into translocation-positive and translocation-negative groups. For cases with translocations, the NF-κB pathway appears to be an important common pathway of activation [124, 130]. This has been shown by mechanistic experiments and gene expression profiling. In contrast, translocation-negative MALT lymphomas overexpress genes involved in inflammation and immune response, such as interleukin-8, CD28, and CD86, among others [130].
Alpha Heavy Chain Disease
This disease is also known as immunoproliferative small intestinal disease and as Mediterranean lymphoma [58, 102]. In alpha heavy chain disease, the neoplastic cells secrete a form of truncated IgA that cannot bind to Ig light chain, as a result of deletions in the VH or CH1 regions of the heavy chain. Morphologically and immunophenotypically alpha heavy chain disease resembles MALT lymphoma, most commonly with extreme plasmacytic differentiation. Patients who live in countries around the Mediterranean Sea or in South Africa are most commonly affected and the disease is associated with poor living conditions. Patients present with diarrhea or malabsorption as a result of disease involving the small intestine and mesenteric lymph nodes. Other gastrointestinal tract sites also can be involved. This disease may respond to antibiotic therapy.
Nodal Marginal Zone Lymphoma
These neoplasms were originally recognized by their cytologic resemblance to reactive monocytoid B-cells, hence the term monocytoid B-cell lymphoma was proposed by Sheibani and colleagues [133]. Subsequently, it became more clear that the neoplastic cells differ from reactive monocytoid cells in their location within the lymph node and immunophenotype. These tumors are currently thought to originate in the lymph node marginal zone and are designated as nodal marginal zone B-cell lymphoma (NMZL) in the WHO classification [134]. The designation as nodal (versus extranodal or splenic) in the WHO system is based on arbitrary clinical criteria. A MZL in lymph node is considered as nodal if there is no evidence of extranodal or splenic disease [134]. NMZL represents less than 2 % of all NHLs.
Patients with NMZL are usually adults in the sixth or seventh decades, with a slight female preponderance [134–136]. The clinical course is indolent and B-type symptoms occur in approximately one-third of patients, similar to patients with other systemic low-grade B-cell NHL. Most patients present with peripheral lymphadenopathy, often initially detected as localized lymph nodes in the head and neck region, but widespread lymphadenopathy is also common. Bone marrow involvement is detected at a variable frequency in different studies, ranging from approximately 30 to 60 %. Hepatitis C infection is associated with cases of NMZL reported from Italy [135]. A serum paraprotein, usually at a low level, and composed of IgG, IgA, or IgM, has been reported in up to one-third of patients [137].
Histologically, lymph nodes involved by NMZL have a pale low power appearance as a result of the abundant cytoplasm of the neoplastic cells. These tumors can show a variety of patterns [135–140]. A diffuse pattern is most frequent, but the NMZLs can colonize follicles imparting a nodular pattern that mimics FL, be interfollicular, or perifollicular. Uninvolved follicles often contain hyperplastic germinal centers. The cytologic features of NMZL are heterogeneous. The most distinctive cell type is the monocytoid lymphocyte which often predominates in cases of NMZL. The tumor cell cytoplasm of monocytoid cells is relatively abundant, pale eosinophilic or clear, with well-delineated cell borders. The tumor cell nuclei are small and cytologically bland. The tumor cell chromatin is relatively clumped and mitotic figures are infrequent. NMZLs also can be composed of centrocyte-like cells or cells with plasmacytoid differentiation. Large cells are also present in variable numbers, but if sheets of large cells are present the neoplasm has transformed to DLBCL.
Immunophenotypic studies have shown that NMZLs are mature B-cell neoplasms that express monotypic Ig light chain common usually IgM, pan-B-cell antigens, CD11c, CD43, and BCL-2 [134, 135]. These tumors usually do not express CD10, CD21, CD23, BCL-6, cyclin D1, and T-cell antigens including CD5. Most cases are negative for IgD but approximately 15 % of NMZLs are positive.
The molecular pathogenesis of NMZL is poorly understood. The Ig genes are rearranged and the Ig variable regions commonly show somatic mutations. Conventional cytogenetic studies have identified a variety of abnormalities, but no consistent abnormalities have been reported. Trisomy 3 has been shown in 50–60 % of cases [131, 141]. Translocations known to occur in MALT lymphoma do not occur in NMZL [142].
Splenic Marginal Zone B-Cell Lymphoma
Splenic marginal zone B-cell lymphomas (SMZL) were so named by Schmid and colleagues [143] because these neoplasms were thought to arise from splenic marginal zone B-cells, and to be closely related to other marginal zone B-cell lymphomas. The cell of origin of SMZL, however, and its relationship to other marginal zone neoplasms is currently uncertain [144].
Patients with SMZL are adults, usually elderly, and have a characteristic disease distribution: spleen, splenic hilar and other abdominal lymph nodes, and bone marrow. Peripheral blood is often involved and the cells commonly have irregular or villous cytoplasmic projections, hence use of an alternative name splenic B-cell lymphoma with villous lymphocytes [145]. Patients present with splenomegaly and laboratory abnormalities, usually anemia or thrombocytopenia, or both [146]. A subset of patients has an associated serum IgM paraprotein and levels can be high [147]. Systemic symptoms are usually absent. Splenomegaly is usually marked, but in a subset of cases the spleen is relatively small and these patients may have early, localized disease.
Grossly, the spleen is usually massive; in one study the splenic weight ranged from 970 to 2,400 g (normal, less than 100–150 g) [148]. Histologically, SMZL preferentially involves the marginal zones surrounding lymphoid follicles of the white pulp and, if more extensive, also replaces lymphoid follicles and extends into red pulp [144–148]. Cytologically, the neoplastic cells have relatively abundant clear cytoplasm and central round, bland nuclei. Plasmacytoid differentiation can be prominent. Mitotic figures are rare and occasional large lymphoid cells are present. Transformation to DLBCL can occur, detected as sheets of large B-cells, and often correlates with a more aggressive clinical course.
Immunophenotypic studies have shown that SMZLs are mature B-cell neoplasms that express monotypic Ig light chain, IgM, usually IgD, pan-B-cell antigens, CD11c and BCL-2. SMZL cells are negative for pan-T-cell antigens, CD10, CD21, CD25, and CD103, and BCL-6 [144, 146, 148]. Most cases of SMZL are negative for CD5 and CD23, but approximately 20 % of cases can be positive for one or both markers. Patients with CD5+ SMZL can have leukemic involvement with high lymphocyte counts raising the differential diagnosis with CLL/SLL [149].
Conventional cytogenetic analysis has shown an abnormal karyotype in up to 75 % of cases [150]. Common abnormalities in SMZLs include gains of chromosome 3, more specifically 3q, gains of 12q, and deletions of 6q and 7q. The latter deletions are most often 7q31–32, present in up to 40 % of cases [144]. Translocations involving the CDK6 gene at chromosome 7q21 have been reported in a small subset of cases [151]. SMZLs contain Ig gene rearrangements. The Ig variable regions genes are commonly mutated [152]. Stereotypy of the Ig genes has been shown in approximately 10 % of cases suggesting that antigen drive is involved in the pathogenesis of this subset [153]. No well-characterized chromosomal translocations occur in cases of SMZL. In particular, translocations that occur in MALT lymphoma are not present in SMZL [142]. Mutations of the p53 and BCL-6 genes have been reported in a subset of cases; p53 mutations correspond with a poorer prognosis [154, 155]. Gene expression profiling of cases of SMZL has shown upregulation of genes involved in intracellular signaling (e.g., AKT1) and transcription as well as genes attributable to normal splenic tissue [156].
Splenic Diffuse Red Pulp B-cell Lymphoma
These neoplasms are low-grade small B-cell neoplasms that diffusely replace the splenic red pulp, unlike SMZLs that preferentially replace splenic white pulp. Based on cytologic and immunophenotypic similarity, the term splenic marginal zone lymphoma, diffuse variant also has been used for these tumors. Splenic diffuse red pulp B-cell lymphoma is not well characterized and also resembles cases of so-called hairy cell leukemia variant. The current WHO classification considers splenic diffuse red pulp B-cell lymphoma and hairy cell leukemia variant to be closely related and groups these neoplasms into a provisional entity designated splenic B-cell lymphoma/leukemia, unclassifiable [157].
Mantle Cell Lymphoma
In the current WHO classification, the definition of MCL is as follows “a B-cell neoplasm generally composed of monomorphic small to medium-sized lymphoid cells with irregular nuclear contours and a CCND1 translocation” [158]. The two constant components of this definition are B-cell lineage and a CCND1 translocation, usually t(11;14)(q13;q32), whereas the morphologic appearance can be variable. Thus, the definition of MCL is essentially a molecular definition and has lead to reclassification of a subset of small B-cell lymphomas that were once classified on the basis of morphology, but are now classified as MCL on the basis of the presence of t(11;14)(q13;q32). MCL represents approximately 6 % of all NHLs [158].
Clinically, patients with MCL are usually elderly and men are affected more often than women, with a male to female ratio of approximately 3 to 1 [159–161]. B type symptoms occur in 30–40 % of patients and 70–80 % of patients present have stage III or IV disease. Most patients present with generalized lymphadenopathy and bone marrow is commonly involved, approximately 60 %. An absolute peripheral blood lymphocytosis of more than 4,000/mm3 is infrequent, but low-level involvement of the blood is common when carefully searched for morphologically or if sensitive molecular techniques are used [162]. Relatively low levels of lymphocytosis probably do not influence survival significantly, but overt leukemia has been associated with a poorer prognosis in some studies [163]. Extranodal disease may occur, usually in association with nodal involvement. Patients may present with numerous polyps involving the gastroesophageal tract below the level of the gastrointestinal junction, also known in the literature as multiple lymphomatous polyposis of the intestine [164]. Involvement of Waldeyer ring can occur. Most patients with MCL have a poor prognosis. In a study by Fisher and associates, after 10 years of clinical follow-up only 8 % of MCL patients were alive after therapy using cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) [165].
Histologically, MCL replaces the lymph node architecture in a diffuse, nodular, or mantle zone pattern (Fig. 42.6). The mantle zone pattern is rare, occurring as a predominant pattern in less than 5 % of cases, and results from the neoplasm selectively involving the follicle mantle zones surrounding germinal centers. A pure mantle zone pattern correlates with a more indolent clinical course [166]. Cytologically, typical cases of MCL are composed of a monotonous population of small lymphoid cells that can have slightly irregular or more pronounced irregular nuclear contours [158, 165, 167]. Large nucleolated lymphoid cells are rare. Other histologic findings common in MCL include numerous eosinophilic epithelioid histiocytes and germinal centers completely surrounded by tumor without a normal lymphoid cuff (so-called naked germinal centers). Plasmacytoid differentiation is rare but can occur in MCL [168].
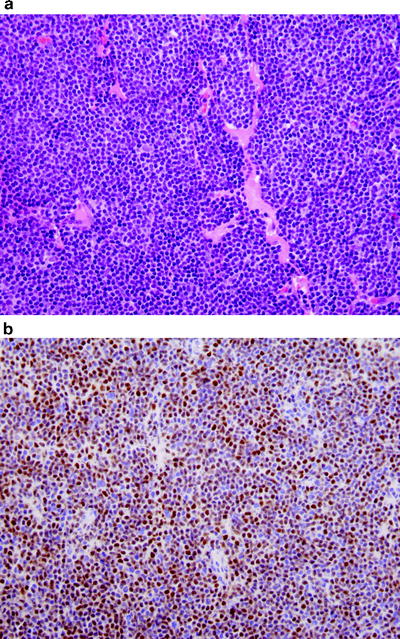
Fig. 42.6
Mantle cell lymphoma involving lymph node. (a) The neoplasm is composed of a monotonous population of small cells. Sclerotic blood vessels, common in mantle cell lymphoma, are also shown. (b) Cyclin D1 immunostain is positive supporting the diagnosis of mantle cell lymphoma. (immunohistochemistry with hematoxylin counterstain, ×400)
Histologically aggressive variants of MCL occur and these can be divided into two types: blastoid and pleomorphic [158]. Blastoid MCLs are characterized by slightly larger lymphoid cells with finely dispersed nuclear chromatin and numerous mitotic features that resemble lymphoblastic lymphoma. Pleomorphic variants of MCL are composed of large cells that resemble, in part, DLBCL. In the blood, there is an aggressive variant of MCL that can resemble B-cell prolymphocytic leukemia [57]. This variant is rare and probably fits within the spectrum of the pleomorphic variant. Histologically aggressive variants of MCL have a more aggressive clinical course. Whether these variants represent transformation from typical cases of MCL or arise de novo is not well established. In patients who have both typical and pleomorphic variant MCL, either simultaneously or sequentially, clonal identity has been shown in most of the few cases analyzed [169, 170].
Immunophenotypic studies have shown that typical cases of MCL express monotypic Ig light chain (more often Ig λ), IgM, IgD, pan-B-cell antigens, BCL-2, and CD5 and are negative for CD3, CD10, CD21, CD23, CD103, MUM1, and BCL-6 [158]. SOX11 has been recently advocated as a new marker helpful for diagnosis of MCL, especially the cyclin D1-negative variant (see text that follows), but SOX11 is not specific [171]. Using flow cytometry immunophenotyping, unlike cases of CLL/SLL, MCLs typically express brighter surface Ig and CD20, and are usually positive for CD79B and FMC-7. Although CD23 is usually negative in MCL, in 10 % of tumors CD23 can be expressed, usually with dim intensity [172]. Cases of MCL can show variant immunophenotypes, but are still recognizable as MCL because they carry t(11;14) and overexpress cyclin D1. Cases of MCL may not express CD5 or can be positive for CD10, BCL-6, or MUM1 [158]. In our experience, variant immunophenotypes are more common in histologically aggressive variants of MCL.
Conventional cytogenetic analysis of MCL cases has shown that the t(11;14)(q13;q32) is present in virtually all cases. This abnormality, however, is present as a sole cytogenetic abnormality in less than 10 % of cases [173]. Usually a number of abnormalities are present in MCL and complex karyotypes (≥3 abnormalities) are very common. Most of these additional abnormalities include deletions and chromosome copy number changes. Deletions of loci at chromosome 9p21 (P16 gene) or 17p (P53 gene) occur relatively often, in up to 20 % of cases, and are found mostly in histologically aggressive variants of MCL. Additional balanced chromosomal translocations are uncommon, but t(8;14)(q24;q32)/MYC-IgH occurs in a small subset of blastoid variants of MCL [174].
The Ig genes are rearranged in MCL. The Ig variable regions are usually unmutated, but approximately 20 % of cases have mutated IgH variable region genes [158]. In the t(11;14)(q13;q32), the CCND1 gene (also known as BCL1) on 11q13 is juxtaposed with the IgH gene on 14q32 resulting in overexpression of cyclin D1. Cyclin D1 facilitates cell cycle transition from G1 to S phase. The breakpoints on chromosome 11q13 are widely scattered over approximately 120 kb. As a result, various methods for detecting the t(11;14) result in markedly different detection rates. PCR methods that currently exist can only assess the major translocation cluster region, involved in approximately 40 % of cases. FISH using large cosmid probes can detect virtually all 11q13 breakpoints and therefore detects virtually all cases with a CCND1 translocation and can be performed on paraffin-embedded tissue sections if necessary [175]. Although conventional cytogenetics should theoretically also detect all cases of the t(11;14), poor growth and sampling error result in a detection rate of 70–80 %, and fresh tissue is required.
MCL has been a subject of intense molecular research in the past decade. A number of high throughput methods have been used to study MCL including gene expression profiling, comparative genomic hybridization, single nucleotide polymorphism (SNP) arrays, microRNA profiling arrays, etc. [176–180]. These methods have shown that MCL cases have a characteristic, relatively homogeneous gene expression profile. Perhaps this is not unexpected since the definition of MCL is, in large part, based on the t(11;14). These studies also highlighted the importance of proliferation in MCL and how this signature can be used to predict prognosis [176]. Subsequent immunohistochemical studies also have shown that Ki-67 or repp86 cell counting (proliferating cells) also can be used to predict prognosis in MCL treated using a variety of therapeutic regimens [161, 181–183]. Other methods have shown that there are numerous chromosomal gains and losses, specific deletions, and mutations in cases of MCL. As has been reviewed elsewhere by Jared and Campo, most of these changes impact the proliferation and cell cycle pathways in the neoplastic cells [184]. Apoptotic mechanisms are also dysregulated in MCL [185].
Cyclin D1-Negative MCL
In the gene expression profiling study by Rosenwald and colleagues [176], a small subset of lymphomas were identified that morphologically and immunophenotypically resembled MCL, but lacked the t(11;14) as well as cyclin D1 expression. A subset of these cases expressed cyclin D2 or cyclin D3 and it was suggested these tumors were truly MCL in which aberrant cyclin D2 or cyclin D3 expression was fulfilling the usual role of cyclin D1, hence the designation as cyclin D1-negative MCL [176, 186]. Subsequent molecular studies have shown that a subset of these cases has chromosomal translocations that involve the cyclin D2 or cyclin D3 locus, analogous to the t(11;14), or share copy number changes with other cases of typical t(11;14) + MCL [184]; indirect evidence to support classifying these cases as MCL. A number of lymphoma types, however, overexpress cyclin D2 or cyclin D3 or both, and some typical MCLs that overexpress cyclin D1 also overexpress cyclin D2 or D3 [187].
What seems to be clear (to this author) is that there are tumors that closely resemble typical cases of MCL at the morphologic and immunophenotypic level and yet lack the t(11;14) and cyclin D1 expression. Should they be designated as MCL or with another name that emphasizes what the tumor is as opposed to what it is not? From the clinical point of view, based on current treatment options, this is currently an academic question, but with the hope of targeted therapy in the future it may be wise to not designate these neoplasms as MCL. In addition, from the pathologic standpoint, the diagnosis of cyclin D1-negative MCL is difficult to establish with confidence without knowledge of the gene expression profile.
Diffuse Large B-Cell Lymphoma
In the current WHO classification, the definition of diffuse large B-cell lymphoma (DLBCL) is based on histologic and immunophenotypic criteria; large B-cells in a diffuse pattern [188]. It is therefore not a surprise that DLBCL is clinically and molecularly heterogeneous. DLBCL most often arises in de novo fashion, but a subset of cases arises via histologic transformation of a low-grade B-cell lymphoma. As result, in large part DLBCL is a “wastebasket.” DLBCL represents up to 40 % of all NHLs [188].
In the 2008 WHO classification, attempts are made to tease out subgroups of DLBCL on the basis of unique histologic features, distinctive immunophenotypes, or relatively unique clinical presentation (e.g., primary DLBCL of the central nervous system). Cases of DLBCL that cannot be further specified into one of these subgroups are designated as DLBCL not otherwise specified (NOS).
In the following, we will discuss DLBCL NOS as well as some of the subgroups of DLBCL that are recognizable as pathologic entities. Clinical variants of DLBCL in which the pathologic findings are those of DLBCL but are otherwise not specific will not be discussed here.
Diffuse Large B-Cell Lymphoma NOS
DLBCL NOS (henceforth specified as DLBCL) occurs mainly in adults, with a median age in the seventh decade [188–191]. Men are affected slightly more often than women. Although less common in children, DLBCLs represent 15–20 % of childhood NHLs. There are differences in immunophenotype and frequency of molecular alterations depending on the geographic region in which DLBCL is being studied.
Patients with DLBCL often have B type symptoms and present with a large, growing mass, often associated with necrosis. Nodal presentation is most common, but extranodal sites are frequently involved, and the disease may be restricted to extranodal sites in up to 40 % of patients. A wide variety of extranodal sites can be involved by DLBCL. Approximately half of patients have stage I or II disease. Bone marrow involvement occurs in approximately 20–30 % of patients [192]. Peripheral blood involvement by large neoplastic cells can be identified in a subset of patients, usually in those patients who have bone marrow disease [188].
If untreated, patients with DLBCL usually die of disease, often within 2 years, however approximately 60 % of patients with DLBCL respond well to chemotherapy and can be cured. For years the mainstay of chemotherapy was the CHOP regimen, but within the past decade rituximab (R) plus CHOP has become the preferred treatment with improvement in overall survival rates [193]. Predicting which patients will respond to therapy, however, remains challenging. In 1994 the International Prognostic Index (IPI) was published [194]. The IPI employs five variables, each of which is worth one point: age >60 years, poor performance status, stage III or IV disease, ≥2 extranodal sites of disease, and above normal serum LDH level. An IPI score of 0–1 indicates good prognosis whereas an IPI score of 4–5 predicts a poor prognosis and the likely need for more aggressive therapy than the R-CHOP regimen. Although very helpful, the components of the IPI are essentially surrogates of DLBCL biology and it is understood that in-depth knowledge of DLBCL pathogenesis will lead to better methods for predicting prognosis as well as identifying specific pathways or molecules for targeted therapy.
Histologically, DLBCLs partially or totally replace normal architecture in a diffuse pattern [188–190]. Less commonly, DLBCL can partially replace organs, and in lymph nodes, DLBCL rarely can be confined to the interfollicular regions or sinuses. The definition of a large cell is based on nuclear size which needs to be greater than that of benign histiocyte nuclei; the latter are virtually always present admixed within lymphomas. Mitotic figures are usually numerous. Cytologically, the neoplastic cells in DLBCL can exhibit a spectrum of findings. The most common cytologic variants are centroblastic, immunoblastic, and anaplastic [188]. Centroblasts are 20–30 μm in size and have round or oval vesicular nuclei with two to three nucleoli and moderate amphophilic cytoplasm. Often one nucleolus is centrally located with one to three additional nucleoli apposed to the nuclear membrane. The term immunoblast refers to an activated lymphocyte. Neoplastic immunoblasts resemble transformed lymphocytes; they are larger than centroblasts with an eccentrically located vesicular round or oval nucleus containing a prominent target-like central nucleolus, and relatively abundant amphophilic cytoplasm (Fig. 42.7). These cells commonly exhibit plasmacytoid differentiation. Anaplastic cells are often very large oval or polygonal cells with pleomorphic nuclei that can resemble, in part, Reed–Sternberg or Hodgkin’s cells.
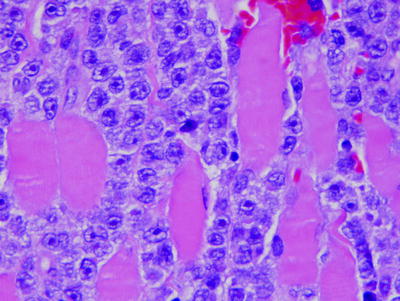
Fig. 42.7
Diffuse large B-cell lymphoma, immunoblastic variant, infiltrating skeletal muscle. (hematoxylin–eosin, ×1,000)
Although these descriptions of centroblasts, immunoblasts, and anaplastic cells are distinctive, it is true that DLBCL can be composed of mixtures of these cells or neoplastic large B-cells can exhibit cytologic features that are intermediate between centroblasts and immunoblasts. Earlier studies of patients with DLBCL showed that patients with the immunoblastic variant had a worse prognosis [195, 196]. However, the problems in reproducibly subcategorizing cases of DLBCL lead the WHO classification to group these variants as DLBCL. In some ways this was an unfortunate decision as cytologic features are most likely a manifestation of gene expression and molecular abnormalities and a subset of DLBCL cases can be designated reliably as centroblastic, immunoblastic, or anaplastic.
Approximately two-thirds of cases of DLBCL express monotypic surface Ig light chain, and IgM is the most common Ig heavy chain expressed [188]. Plasmacytoid DLBCLs commonly express monotypic cytoplasmic Ig. Approximately one-third of DLBCLs are Ig negative. DLBCLs express pan-B-cell antigens, 60–70 % express BCL-2, and a subset is positive for CD10, CD23, BCL-6, or MUM1 [188–191, 197]. DLBCLs are usually negative for T-cell antigens, but approximately 10 % of cases can be CD5+. Patients with CD5+ DLBCL are reported to have more aggressive disease [198]. Most cases of DLBCL express activation markers and have a substantial proliferation rate. Major histocompatability complex (MHC) class I or II antigens can be absent in a subset of DLBCLs; these cases are reported to have a poorer prognosis [188]. CD30 is expressed by a subset of DLBCL and in most cases of the anaplastic variant.
Cytogenetics and molecular studies have shown that approximately 50 % of DLBCLs carry chromosomal translocations. The most common translocation, in 20–30 % of cases, involves chromosome 3q27, the site of the BCL-6 gene [188, 189]. The most common specific translocation is t(3;14)(q27;q32) but there are many others partners of BCL-6 that can be involved in translocations. Another subset of 10–20 % of DLBCL cases carries the t(14;18)(q32;q21) involving the BCL-2 and IgH genes. Clinically, patients with de novo DLBCL and BCL-2 rearrangement have an increased tendency to relapse compared with patients with DLBCL without BCL-2 rearrangement, more akin to the behavior of follicular lymphomas [199]. The MYC oncogene is stated to be rearranged in approximately 10 % of DLBCLs [188]. In our experience, MYC rearrangements are more common in cases with evidence of high proliferation, such as a starry sky pattern or high numbers of Ki-67+ cells. MYC rearrangements may involve Ig partners, most often t(8;14)(q24;q32) involving IgH, or non-Ig partners. MYC rearrangements predict a poorer prognosis. Other common cytogenetic abnormalities in DLBCL include gains of chromosomes 3q, 12q12, and 18q21–22 and losses of 6q21–22 [200, 201].
The Ig genes are rearranged in DLBCL and the Ig variable region genes are commonly mutated [188]. P53 gene mutations occur in approximately 15 % of DLBCLs and predict prognosis. In particular, mutations located specifically in the DNA binding domain of P53 are associated with the worst prognosis [202]. The BCL-2 gene is amplified in approximately 20 % of cases. Deletions in the FAS gene death domain are reported in approximately 20 % of cases, resulting in defects in the extrinsic apoptotic pathway. Other genes that can be mutated in DLBCL with some frequency include BCL-6, MYC, PAX5, PIM1, and P16 [203].
A number of high throughput methods have been used to study DLBCL. It is probably not an exaggeration to conclude that gene expression profiling using cDNA microarrays has made its greatest impact in DLBCL. These studies have shown that DLBCL is heterogeneous and can be subdivided into prognostic subgroups [204, 205]. The most popular system divides DLBCL cases into germinal center B-cell like (GCB) and activated B-cell-like (ABC) groups [205]. Patients with GCB tumors have a better prognosis, independent of the IPI, in patients treated with either CHOP or R-CHOP chemotherapy regimens. Most of the cases of DLBCL with t(14;18) fall within the GCB group [206]. NF-kappa B pathway activation has been found to be more prominent in the ABC type [207]. Chronic active B-cell receptor signaling also has been shown in the ABC type [208]. It should be noted, however, that a subset of DLBCLs cannot be classified as either GCB or ABC type, up to 30 % in some studies.
As gene expression is not yet readily available for routine diagnosis, a number of groups have suggested various immunohistochemical staining algorithms to predict the GCB versus ABC types of DLBCL. The best known system is the so-called Hans classifier which has an accuracy of 76 % [209]. A recently proposed system developed by Choi and colleagues uses 8 antibodies and is stated to predict the gene expression type correctly in approximately 90 % of cases of DLBCL [210].
More recent gene expression studies have proposed additional gene signatures that correlate with better or worse prognosis. In the study by Lernz et al., a so-called stromal-1 signature related to extracellular matrix and histiocyte infiltration predicted a better survival whereas a stromal-2 signature attributable to blood vessel density correlated with poorer survival [211]. Monti and colleagues have shown three gene signatures in DLBCL: oxidative phosphorylation, B-cell receptor/proliferation, and host response [212]. These signatures show important correlations with genetic data. Cases of DLBCL with the oxidative phosphorylation signature commonly carry t(14;18) or FAS death domain deletions. DLBCLs with the B-cell receptor/proliferation signature commonly have BCL-6 abnormalities and the host response gene signature includes all cases of T-cell/histiocyte rich large B-cell lymphoma.
A subset of cases of DLBCL carry the t(14;18)(q32;q21) resulting in BCL-2 overexpression and also carry MYC gene rearrangements. These tumors have been referred to as “double hit” or MYC/BCL-2 or M/B DLBCLs [213–215]. Patients with these neoplasms have a very poor prognosis. These tumors may be included in the category of DLBCL or may be grouped in the “gray zone” category of B-cell lymphoma, unclassified, with features intermediate between DLBCL and Burkitt lymphoma [188]. In our opinion, these MYC/BCL-2 DLBCLs need to be included in the next version of the WHO classification as a separate entity.
T-Cell/Histiocyte Rich Large B-Cell Lymphoma
T-cell/histiocyte rich large B-cell lymphoma (TCHRLBCL) is a histologically distinctive type of DLBCL in which the neoplastic large B-cells are few and are associated with numerous reactive T-cells and histiocytes in the biopsy specimen. No cutoff for the number of large B-cells is provided in the WHO classification except that the B-cells should not be present in aggregates or sheets, as these would support classification as DLBCL NOS [216]. Approximately 300 cases of TCHRLBCL have been reported in the literature.
Patients with TCHRLBCL tend to be adults with a slight male predominance [217–219]. Patients often have B-type symptoms and advanced stage disease involving lymph nodes, bone marrow, liver, and spleen. Patients are often refractory to therapy, and the disease can resemble DLBCL NOS in relapse biopsy specimens. Some cases of TCHRLBCL may arise from underlying nodular lymphocyte predominant Hodgkin’s lymphoma.
Histologically, TCHRLBCLs have a diffuse pattern and are composed predominantly of small reactive T-cells and benign nonepithelioid histiocytes [216, 217]. Granulocytes and plasma cells are absent. Usually <5–10 % of all cells are singly scattered large, neoplastic B-cells than can appear as centroblasts, immunoblasts, pleomorphic Reed–Sternberg-like cells, or lymphocyte predominant (LP)-like large cells.
Immunohistochemical studies have shown that the large cells express pan-B-cell antigens, often express BCL-2 and BCL-6, and are negative for CD15 and CD138 [216]. The small lymphocytes and histiocytes express T-cell and histiocyte-associated markers, respectively. CD30 can be expressed in a subset of cases.
The large B-cells in TCHRLBCL carry Ig gene rearrangements and the Ig variable regions genes are commonly mutated. Comparative genomic hybridization has shown some genetic imbalances, most commonly gains involving chromosome loci Xq, Xp21q11, 4q13q28, and 18q21, and loss of 17p [220]. Gene expression profiling studies have shown that these tumors have a host response signature, which is in keeping with the morphologic and immunophenotypic findings [212, 221].
Lymphomatoid Granulomatosis
Lymphomatoid granulomatosis (LyG), originally described by Liebow and colleagues, was originally thought to have features that overlapped with both malignant lymphoma and Wegener’s granulomatosis, leading to its name [222]. LyG is now considered to be neoplastic at onset, is histologically distinctive, is associated with EBV infection, and therefore is included as a type of DLBCL in the WHO classification [223]. LyG is a rare disease.
Patients with LyG are usually middle aged adults and there is a male predominance. There appears to be an increased frequency of LyG in immunodeficient patients. Patients present with pulmonary and systemic symptoms and chest radiographs typically show discrete, round masses, most often bilateral [222]. Other sites that can be involved frequently by LyG include the kidney, liver, central nervous system, and skin. The upper aerodigestive tract is rarely involved [223].
Histologically, LyG is characterized by an angiocentric and angiodestructive infiltrate of lymphocytes, plasma cells, and histiocytes [223–225]. Granulocytes are rare. Necrosis can be prominent. In low-grade cases of LyG, few large B-cells are present whereas in high-grade cases aggregates or sheets of large B-cells are present. The natural history of LyG is to accrue large cells and evolve into a process that resembles DLBCL NOS [222].
Immunophenotypic studies of LyG have shown that the many or most of the lymphoid cells in biopsy specimens are small reactive T-cells. The large cells, however, which represent the malignant cell population, express pan-B-cell antigens and commonly carry EBV. The large B-cells can be CD30+, but are always CD15−. Polymerase chain reaction studies have shown monoclonal Ig gene rearrangements and monoclonal EBV episomal DNA in most cases [223].
Primary Mediastinal Large B-Cell Lymphoma
The WHO classification defines primary mediastinal large B-cell lymphoma (PMLBL) as a DLBCL arising in the mediastinum of putative thymic B-cell origin with distinctive clinicopathologic, immunophenotypic, and molecular features [226]. Applying this definition, however, is not always as easy as it seems clear. A major problem is the difficulty in determining that a neoplasm arises in the mediastinum and/or the thymus. The mediastinum is rich in lymph nodes that can be involved by systemic DLBCL. Therefore, the category is somewhat “dirty” as it is difficult to exclude all cases of nodal DLBCL that present with a mediastinal mass from the category of PMLBL. The problem is further exacerbated by the increased use of fine needle aspiration and needle core biopsy for diagnosis of mediastinal masses. Although these specimens allow the diagnosis of DLBCL, their small size precludes or makes more difficult the observation of certain features highly suggestive of PMLBL. Despite these limitations, the category of PMLBL has unique clinicopathologic, immunophenotypic, and molecular features [226].
Patients with PMLBL are most often young adults, with onset in the fourth decade [227–229]. There is a female predominance with a male/female ratio of 1 to 2. Patients typically present with a large, bulky (>10 cm) mass that causes local symptoms such as cough, chest pain, and shortness of breath. Approximately two-thirds of patients present with localized disease and approximately one-third of patients may develop superior vena cava syndrome. Patients typically have high serum levels of LDH but low serum levels of beta-2-microglobulin. Bone marrow involvement or dissemination of disease at time of diagnosis is absent as, by definition, their presence supports systemic disease [226]. Admittedly, this is a clinical definition that is somewhat arbitrary as patients with PMLBL can theoretically disseminate before first diagnosis of disease. Patients respond poorly to conventional chemotherapy (such as CHOP) but others have a relatively good prognosis if patients are aggressively treated with combination chemotherapy and irradiation [227–229]. Relapses commonly occur at extranodal sites, most commonly kidney, liver, adrenal gland, and brain [226].
Histologically, PMLBL diffusely replaces the thymus and invades into contiguous structures including the lungs, pleura, pericardium, and can spread to regional lymph nodes (Fig. 42.8). PMLBL is composed of large lymphoid cells that exhibit a spectrum of cytologic appearances: most commonly resembling centroblasts, and less often resembling immunoblasts, or Reed–Sternberg-like cells [226, 230, 231]. The neoplastic cells often have abundant pale cytoplasm (so-called clear cells). Sclerosis is common and often compartmentalizes the neoplastic cells into groups. Mitotic figures are usually numerous.
Fig. 42.8
Primary mediastinal (thymic) large B-cell lymphoma. Residual thymus gland is present at the right of the field (hematoxylin–eosin, ×20)
Immunophenotypic studies have shown that the neoplastic cells express pan-B-cell antigens and B-cell transcription factors (PAX5, BOB.1, OCT-2, and PU.1) but are commonly negative for surface Ig [232, 233]. Cyclin E and MAL are commonly overexpressed [233, 234]. Approximately 75 % of cases are weakly and/or heterogeneously positive for CD30 and large subsets of tumors are positive for MUM1, CD23, BCL-2, or BCL-6 [226]. CD10 is expressed less often, in up to one-third of cases. T-cell antigens including CD5 are negative as is CD21. PMLBLs show immunophenotypic evidence of NF-kappa B activation: TRAF1 and nuclear REL. A subset of cases lack MHC class I or II antigens [226].
PMLBLs carry Ig gene rearrangements and the Ig variable region genes are commonly mutated. Balanced chromosomal translocations are uncommon. Comparative genomic hybridization has shown gains at chromosomal loci 2p15, 9q24, Xp11.4-21, and Xq24–26 and losses at 1p13, 4q, 6 g, 7p, and 17p [235]. A number of genes are amplified in subsets of PMLBL including JAK2, REL, BCL-11A, and others. The BCL-6 or SOCS1 genes are commonly mutated in PMLBL [232, 236]. Gene expression profiling has shown that PMLBL has a signature distinct from GCB- or ABC-like DLBCLs, sharing approximately one-third of its gene expression profile with classical Hodgkin’s lymphoma [237]. Both the NF-kappa B and JAK-STAT pathways are activated in PMLBCL, and these tumors also have a host response gene signature as described previously [238].
Intravascular Large B-Cell Lymphoma
Intravascular large B-cell lymphoma, also known as angiotropic lymphoma and originally described as malignant angioendotheliomatosis, is a rare variant of DLBCL in which the neoplastic cells grow selectively within blood vessels, especially capillaries [239]. Although the histologic pattern of IVLBCL is quite distinctive, the clinical presentation and immunophenotypes of IVLBCL are strikingly heterogeneous, suggesting that more than one etiology or cell of origin is involved. Rare cases of IVLBCL of T-cell or natural killer cell lineage are also reported that are not included in this category, but further highlight the heterogeneity of intravascular lymphomas. The explanation for this pattern of infiltration in IVLBCL is unknown, but the neoplastic cells have been shown to express various chemokines (e.g., CXCR4) or to not express certain types of homing receptors [239, 240].
Patients with IVLBL are typically middle aged or elderly, with a slight male predominance, and most patients have B-type symptoms [241, 242]. There are differences in clinical presentation dependent on geographical location. Patients in Western countries tend to present with symptoms attributable to specific organs involved by lymphoma. In Asian countries, by contrast, patients often present with widespread disease dissemination, hepatosplenomegaly, and can have cytopenias as a result of hemophagocytic syndrome [239, 242]. Organs commonly involved by IVLBCL include the central nervous system, skin, and kidney but any site may be involved. Standard radiographic studies often falsely do not detect disseminated disease, as no masses are present, but IVLBCL can be detected by FDG-PET scan [243]. Bone marrow involvement, although difficult to appreciate morphologically, is relatively common when the bone marrow is assessed using immunophenotypic or molecular methods [244, 245]. IVLBCL tends to not involve lymph nodes. The prognosis is often poor due to late detection of disease and poor response to chemotherapy [239, 241].
Histologically, IVLBL is characterized by the presence of large lymphoid cells filling and/or distending vascular lumina [239, 241]. The neoplastic cells are found primarily within capillaries or small blood vessels. Cytologically, the neoplastic cells can resemble centroblasts or immunoblasts. The tumor cells are often associated with tissue hemorrhage or necrosis and fibrin thrombi can be observed. Extravascular involvement can occur in patients with IVLBCL, and although not prominent clinically or easily detected radiographically, is not uncommon at autopsy.
With the advent of immunophenotypic studies, IVLBL was conclusively shown to be a variant of large B-cell lymphoma. These tumors express pan-B-cell antigens and a subset expresses monotypic Ig [239, 241]. These tumors commonly express MUM1, approximately 30–40 % of cases are CD5+, and a smaller subset can express CD10. BCL-6 is usually negative. Proliferation rate, as shown by Ki-67 immunostaining, is usually high. A case report described an apparently unusual case of IVLBCL that was positive for human herpes virus type 8 [246].
Molecular studies have shown Ig gene rearrangements in IVLBL. Rare cases assessed by conventional cytogenetics have shown a complex karyotype.
ALK + Diffuse Large B-cell Lymphoma
This is a pathologically and immunophenotypically distinct type of DLBCL in which the neoplastic cells resemble immunoblasts or plasmablasts and express anaplastic lymphoma kinase (ALK) [247]. ALK+ DLBCL is a truly rare disease representing <1 % of DLBCL cases.
ALK+ DLBCL can affect patients of all ages with a male to female ratio of 3 to 1 [247, 248]. Lymph nodes are commonly involved and patients tend to present with advanced stage disease, often with involvement of extranodal sites. Histologically, ALK+ DLBCL can replace lymph nodes diffusely or exhibit a sinusoidal pattern of involvement or both. The neoplastic cells are large with prominent central nucleoli and abundant cytoplasm resembling either immunoblasts or plasmablasts. The neoplastic cells are usually positive for plasma cell-associated markers (e.g., CD38, CD138), cytoplasmic IgA, and EMA, and can express CD4, CD43 (often focal), CD45 (usually dim if present), CD57, CD79a (often bright), PAX5 (often negative) and MUM1, and are negative for CD10, CD20, CD30, BCL-6, T-cell antigens, and EBV. The proliferation rate, shown by Ki-67, is often brisk.
The most characteristic feature of ALK+ DLBCL is the presence of ALK expression. ALK is expressed as a result of molecular abnormalities involving the ALK gene at chromosome 2p23. The most common abnormality is t(2;17)(p23;q23) involving the ALK and clathrin genes [247, 249]. Less commonly the t(2;5)(p23;q35) is present in these tumors. The ALK immunostaining pattern correlates nicely with these abnormalities, being cytoplasmic and flocculent in cases with t(2;17) and both nuclear and cytoplasmic in cases with t(2;5). This is attributable to the normal cellular location of the clathrin and nucleophosmin proteins, in cytoplasmic vesicles or the nucleus, respectively [247]. Rare cases are reported with other abnormalities of the ALK gene locus [249, 250].
Plasmablastic Lymphoma
Plasmablastic lymphoma (PBL) is defined in the WHO classification as a diffuse, monomorphous proliferation of large neoplastic cells that resemble B-immunoblasts but have an immunophenotype of immature plasma cells [251]. PBL is a rare neoplasm that most often occurs in patients with immunodeficiency, most commonly human immunodeficiency virus (HIV) infection. With the immunophenotype of PBL being virtually identical to that of plasma cell myeloma, the inclusion of PBL as a rare type of DLBCL has been questioned [252]. Patients with PBL, however, have a clinically aggressive tumor and DLBCL-type therapy for patients with PBL may be appropriate.
This neoplasm was originally described by Delecluse and colleagues [253] in the oral cavity of patients with HIV infection, but it is now recognized that PBL can occur in virtually any site [251]. Extranodal sites are most often involved. Most patients with PBL have HIV infection, but these tumors have been reported in patients with other types of acquired as well as congenital immunodeficiency. A small subset of patients without known immunodeficiency also can develop PBL. PBL most often arises in adults with a male predominance. Many patients have high stage disease and a high IPI score. The prognosis of patients with PBL is poor, despite therapy, with a median survival of approximately 1 year [254].
Histologically, PBL is composed of large neoplastic cells arranged in a diffuse pattern. A starry-sky pattern and a high mitotic rate are common. The neoplastic cells have vesicular nuclei with single prominent central nucleoli and abundant cytoplasm with variable plasmacytoid differentiation. Immunophenotypically, the neoplastic cells are positive for CD38, CD138, and MUM1 and are usually negative for the CD20 and PAX5, although weak expression by a subset of cells can occur in a small subset of cases. The cells of PBL are also commonly positive for cytoplasmic Ig (usually IgG), CD79A, CD30, EMA, and p53 and can express CD10, CD56, and CD4. PBLs are usually negative for CD45/LCA, CD3, CD5, and HHV-8. BLIMP1 and XBP1 have been recently suggested as useful markers for the diagnosis of PBL [255]. The proliferation rate, assessed by Ki-67 expression, is usually high. Approximately 75 % of cases express EBV small encoded RNA (EBER), but not EBV latent membrane protein.
PBLs carry monoclonal Ig gene rearrangements and the Ig variable regions genes are often mutated [251]. MYC is rearranged in a subset of cases [256]. One study has used comparative genomic hybridization to compare DLBCL with PBL cases. The most common chromosomal gains in PBL involved 1p, 1q, and 7q, similar to cases of DLBCL, and used to support the opinion that PBL is best classified as a type of DLBCL [257]. However, the authors did observe differences in segmental chromosomal gains and losses between PBL and DLBCL that the authors seemed to minimize. It seems that the classification of PBL, and its relationship with DLBCL, needs additional study although the clinically aggressive nature of PBL is not in doubt.
Primary Effusion Lymphoma
Primary effusion lymphoma (PEL), also known as body cavity-based lymphoma, is a very rare neoplasm of large B-cells that usually involves one of the body cavities: pleural, pericardial, or peritoneal [258]. Usually, only one body cavity is involved. In a subset of patients, PEL also can involve tissue sites as a solid mass and rarely the tumor can initially present as an extracavitary mass. Extracavitary sites are usually extranodal, but we have seen cases of PEL initially diagnosed by lymph node biopsy. Almost all cases of PEL arise in the setting of immunodeficiency, most commonly HIV infection. Rarely, PEL has been reported in patients who have undergone organ transplantation or in elderly patients without obvious evidence of immunodeficiency [258]. The prognosis of patients with PEL is poor, with overall survival commonly less than 1 year.
Morphogically, the neoplastic cells resemble immunoblasts or plasmablasts with prominent nucleoli and abundant basophilic cytoplasm [258, 259]. In some cases, anaplastic or Reed–Sternberg-like cells are present. The tumor cells are found within body cavity fluids, but may adhere to and invade body cavity surfaces. In lymph nodes, PEL has a sinusoidal pattern of infiltration. Immunophenotypic studies have shown that PEL cells express CD30, CD38, CD138, EMA, and HLA-DR and are usually negative for Ig (surface and cytoplasmic), pan-B-cell antigens (e.g., CD19 and CD20), and BCL-6 [258, 259]. All cases are positive for HHV-8 and most cases are positive for EBER and CD45/LCA. A subset of PEL cases can express T-cell associated antigens. Rare cases of “PEL” are reported that lack evidence of HHV-8; whether these cases are truly PEL or clinicopathologic mimics is uncertain [260].
Molecular studies have shown monoclonal Ig gene rearrangements and a subset of cases also has T-cell receptor gene rearrangements (lineage infidelity) [258]. MYC translocations have been reported rarely [260]. Comparative genomic hybridization has shown frequent gains of chromosome 12p [261]. One gene expression profiling study showed a common gene signature in PEL cases that was distinct from other types of NHL including HIV-associated NHLs. This signature shares features with EBV + transformed lymphoblastoid and plasma cell myeloma cell lines [262].
Burkitt Lymphoma
Clinically, Burkitt lymphoma (BL) cases can be divided into three groups: endemic, sporadic, and immunodeficiency associated. BL also can present in leukemic phase with preferential involvement of peripheral blood and bone marrow.
Endemic BL was first described by Denis Burkitt in equatorial Africa (Uganda) where it occurs within 15° latitude north or south of the equator [263]. Other endemic areas include Papua, New Guinea and Northern Brazil. Evidence of EBV infection is present in over 95 % of patients. There is also a link with malaria and arbovirus infection [264]. The median age of patients with endemic BL is in the range of 4–7 years, with a boy/girl ratio of 2 to 1. The jaw is the most well-known site of disease, involving either the maxilla or mandible in 50–60 % of patients, but large abdominal masses involving retroperitoneal structures, the gastrointestinal tract, or the gonads are also commonly present [263, 265].
Sporadic BL occurs in industrialized nations and represents approximately 1 % of all NHLs in adults [265, 266]. In children, sporadic BL is the most common type of NHL, up to 40–50 % of all cases of NHL. Approximately 20 % of patients have evidence of EBV infection. Patients are usually in the second or third decades of life, with a male/female ratio of 2–3 to 1. The jaw is infrequently involved and most patients present with large abdominal masses, frequently involving the ileocecal region of the gastrointestinal tract. Bone marrow and central nervous system involvement are uncommon at time of presentation, in approximately 10–20 % of cases, but are frequent sites of disease later in the clinical course.
Burkitt lymphoma also occurs in the clinical setting of immunodeficiency, most often HIV infection. Immunodeficiency-associated BL is associated with EBV infection in 30–40 % of cases, and most often involves extranodal sites [265, 267]. As is the case for all types of BL, EBV within tumor cells is episomal, consistent with latent infection, often present in multiple copies per cell, and monoclonal, indicating that the virus is present prior to neoplastic transformation. However, as is evident by the lower frequency of EBV in sporadic and immunodeficiency BL, EBV alone is not sufficient to cause BL.
As BL is most often extranodal, and patients commonly present with a high tumor burden and bulky disease, traditional staging systems (e.g., Ann Arbor system) are of less prognostic value than schemes that assess tumor burden and amenability to surgical excision [268]. Patients with BL have a high risk of central nervous system involvement. Waldeyer ring and the mediastinum are involved uncommonly [265]. Since BL is often widely distributed at time of presentation and has an extremely rapid clinical course, systemic chemotherapy is the treatment of choice [265, 269]. Patients with BL respond dramatically to combination chemotherapy regimens; over 75 % of patients (80–90 % of children) including those who have high-stage disease respond completely with long-term survival [269]. Patients with immunodeficiency-associated BL also respond well to combination chemotherapy, providing their underlying immunodeficiency syndrome allows adequate therapy to be administered.
Histologically, the endemic, sporadic, and HIV-associated types of BL are very similar and are characterized by two essential features: the neoplastic cells have nuclei that approximate the size of the nuclei of benign histiocytes and the tumor has extraordinarily high mitotic and proliferation rates [265, 267]. At low power, the neoplasms grow as expansile masses that diffusely infiltrate contiguous tissues. Reactive histiocytes are scattered throughout the tumor. The relatively clear cytoplasm of the histiocytes in a background of blue neoplastic cells imparts a “starry-sky” appearance (Fig. 42.9). This pattern results from rapid cell turnover with individual cell necrosis and scavenging of debris by macrophages. The neoplastic cells are round to ovoid, strikingly monotonous, and uniform in shape. The nuclear membrane is prominent, and the chromatin is coarse, with two to five prominent, basophilic nucleoli. Mitotic figures are numerous. Although the growth pattern of the tumor is usually diffuse, occasionally selective involvement of germinal centers in partially effaced lymph nodes imparts a nodular pattern.
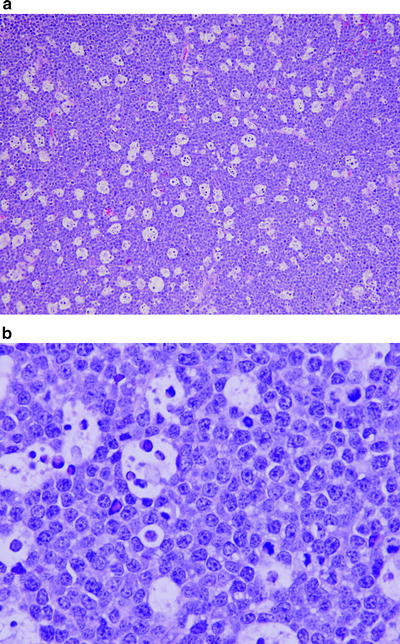
Fig. 42.9
Burkitt lymphoma. (a) Low magnification shows the characteristic starry sky pattern with the histiocytes appearing as clear spaces or “stars” in a background “sky” of darker lymphoma cells. (b) The lymphoma cells are of intermediate size with multiple small nucleoli (hematoxylin–eosin, a. ×200; b. ×1,000)
Immunophenotypic studies of endemic, sporadic, and AIDS-associated BLs have shown similar findings. These tumors are of mature B-cell lineage and typically express monotypic Ig light chain, IgM, pan-B-cell antigens, CD10, CD38, BCL-6, and SOX11 [270]. These tumors are usually negative for IgD, CD21, CD23, CD25, T-cell antigens, TdT and BCL-2 [265]. Immunophenotypic variants have been reported, however, with a subset of cases MUM1+ and minor subsets negative for CD10, BCL-6, or Ig, or weakly positive for BCL-2 [265, 271, 272].
Approximately 80 % of cases carry the t(8;14)(q24;q32), with the remaining cases having one of two variant translocations: 15 % carry t(2;8)(p11;q24) and 5 % carry t(8;22)(q24;q11). Common to each of these translocations is involvement of chromosome region 8q24, the site of the MYC gene. Via these translocations, MYC is juxtaposed with the IgH on the derivative chromosome 14, or the Igκ and Igλ genes are juxtaposed with MYC on the derivative chromosome 8 [273]. The MYC gene has a central role in normal cellular proliferation. This juxtaposition of MYC and Ig gene enhancers results in deregulation of the MYC gene, with increased MYC protein driving cell proliferation [274].
MYC mutations also occur in BL and may enhance tumorigenicity.
Burkitt lymphomas have Ig gene rearrangements and Ig variable region genes are commonly mutated. Additional cytogenetic abnormalities also occur in BL. In one study, abnormalities of chromosome 12q and 22q were associated with worse prognosis in children, and abnormalities of chromosome 17 correlated with worse prognosis in adults. There was no significant difference in karyotype complexity between children and adults with BL [275]. In general, the karyotypes of BL are simpler than those observed in DLBCL or gray zone lymphomas (discussed in text that follows) [276].
The breakpoints of the t(8;14) are distinctive in endemic and sporadic BL [273]. In endemic BL, the breakpoint on chromosome 8 occurs far 5′ to the location of the MYC gene whereas the breakpoint on chromosome 14 usually occurs within the joining region of the IgH gene. By contrast, in sporadic BL, the breakpoint on chromosome 8 occurs within the MYC gene, usually in the first exon, or in adjacent flanking sequences. The breakpoint on chromosome 14 may occur within the IgH switch regions. These differences suggest endemic and sporadic BL arise at different stages of B-cell differentiation and may derive from mistakes that occur as a result of somatic mutation or Ig rearrangement, respectively.
Two major studies of BL using gene expression profiling were published in 2006 [277, 278]. These studies, which focused on sporadic BL, showed that BL has a distinctive gene expression profile and that this profile was identified in a small subset of cases that lacked Ig-MYC translocations as well as a small subset of cases that resembled DLBCL. In the study by Hummel and colleagues [277], the authors emphasized that there is a spectrum of gene expression that occurs between DLBCL and BL. In both studies, rare lymphomas that carried both Ig-MYC and t(14;18)(q32;q21)/IgH-BCL-2 translocations were identified that had a gene expression profile of BL and yet did not behave as do most patients with BL [277, 278]. In retrospect, these “double hit” lymphomas should not have been included in the BL category. MYC also regulates microRNA expression, including up to 60 microRNAs [274]. This area of research is likely to further clarify the pathogenesis of BL.
B-Cell Lymphoma, Unclassifiable, with Features Intermediate Between Diffuse Large B-cell Lymphoma and Burkitt Lymphoma
Although the term B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma (BCLU) suggests a new entity, in fact, there is great overlap between the tumors in this category and lymphomas designated previously in the International Working Formulation as small noncleaved cell lymphoma, non-Burkitt type, or in the REAL classification as high-grade B-cell lymphoma, Burkitt-like. The definition of BCLU is vague in the WHO classification, perhaps not surprising as the definition attempts to define a “gray zone” category [279]. The WHO book states that this is a heterogeneous category “that is not considered a distinct disease entity” but this has not stopped others from writing about BCLU as a lymphoma type.
Morphologically, many cases of BCLU have histologic features that are intermediate between DLBCL and BL [279, 280]. Histologically, these neoplasms are diffuse, often have a starry-sky pattern, and have a high proliferation rate (>99 %) with numerous mitotic figures. The neoplastic cells, however, are of intermediate size similar to BL cells, but with greater nuclear pleomorphism, or are larger with more vesicular nuclear chromatin somewhat similar to DLBCL cells, but not as large.
Cases of BCLU can histologically resemble BL but have atypical immunophenotypic features [279, 280]. For example, these tumors express pan-B-cell antigens, CD10 and BCL-6 with have a high proliferation (Ki-67) rate, and yet also strongly positive for BCL-2. Epstein–Barr virus is uncommon in BCLU.
Cases of BCLU also can have histologic and immunophenotypic features similar (but not identical) to BL but have distinctive cytogenetic or molecular findings. MYC translocations are present in 40–50 % of cases. These translocations often do not use the Ig genes as partners [279–281]. The karyotype of cases of BCLU is often complex. This group also includes so-called double hit or triple hit lymphomas, with simultaneous presence of MYC and BCL-2, MYC and BCL-6 or MYC, BCL-2, and BCL-6 translocations.
The optimal therapy for patients with BCLU is unclear as studies on patient populations with BCLU defined according to the WHO classification are few at this time MYC translocations correlate with a poorer prognosis. These tumors are known to be clinically aggressive and the Hyper-CVAD regimen is often used at our institution.
B-cell Lymphoma, Unclassifiable, with Features Intermediate Between Diffuse Large B-cell Lymphoma and Classical Hodgkin’s Lymphoma
This is another type of “gray zone lymphoma,” the definition of which is also vague in the WHO classification: these neoplasms have “overlapping clinical, morphological, and/or immunophenotypic features between classical Hodgkin’s lymphoma and DLBCL” [282].
Most cases designated as B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin’s lymphoma CHL are tumors at the interface between primary mediastinal large B-cell lymphoma (PMLBL) and classical Hodgkin’s lymphoma (CHL). As a result, patients tend to be 20–40 years of age and they present with a large anterior mediastinal mass, with or without supraclavicular lymph nodes. The clinical course of affected patients is thought to be more aggressive than that of patients with either PMLBL or CHL. There is no consensus for an optimal approach to therapy at this time [282].
Using histologic and immunophenotypic findings, there are essentially three variants of B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL. In one variant, the neoplasm is composed of sheets of large neoplastic cells resembling PMLBL, but the immunophenotype has features more in keeping with CHL (e.g., CD15+ or EBV+). In another variant, the neoplasm resembles CHL, usually nodular sclerosis type, but the neoplastic cells have an immunophenotype more like DLBCL (e.g., LCA+, strong expression of B-cell antigens). A third variant, most common, has mixed features. A subset of these tumors expresses MAL, as is seen in PMLBL. Strong CD20 expression is acceptable in cases diagnosed as CHL, and by itself, is not a reason to classify the neoplasm as a gray zone tumor.
Gene expression profiling of cases of PMBCL and CHL has shown that these neoplasms share aspects of their gene expression profiles [237, 238]. Approximately one-third of the genes expressed in PMLBL and CHL are shared. These data provide, in part, a rationale for the WHO proposal of this category of disease. It seems likely that this category is likely to evolve substantially in the future.
T-Cell Non-Hodgkin’s Lymphomas
The classification of T-cell lymphomas and leukemias is, in many ways, currently more challenging than B-cell lymphomas. This is because T-cell NHLs do not seem to correspond to stages of differentiation, nor are most T-cell NHLs characterized by distinct chromosomal translocations, two key features useful for the classification of B-cell NHLs. As a result, the classification of some types of T-cell lymphoma, particularly extranodal forms, currently incorporates clinical findings into the classification system [14]. Knowledge of T-cell function, likely to emerge over time, will be helpful to more specifically classify T-cell lymphomas and leukemias. In the following sections, the focus is placed on T-cell neoplasms that usually or commonly present as lymphoma.
T Lymphoblastic Leukemia/Lymphoma
T lymphoblastic leukemia/lymphoma can present in two fashions: (1) as acute lymphoblastic leukemia (T-ALL) with extensive involvement of bone marrow and blood; and (2) as lymphoma (T-LBL), most often as an anterior mediastinal mass or lymphadenopathy. Patients who present as T-LBL often convert to a T-ALL clinical picture over time, and many patients who present as T-ALL have an anterior mediastinal mass. This clinical overlap, as well as overlap in immunophenotypic and molecular features, led the authors of the WHO classification to combine T-ALL and T-LBL cases into one category [283]. There are some immunophenotypic and molecular differences between T-ALL and T-LBL, however, that may explain their different clinical presentations [284]. Here we focus on patients presenting as T-LBL.
Patients with T-LBL are most often adolescents and young adults with a male to female ratio of 2 to 1 [283, 284]. Patients commonly present with widespread, supradiaphragmatic lymphadenopathy and/or an anterior mediastinal mass. Patients with a mediastinal mass may have dyspnea, cough, stridor, or superior vena cava syndrome. Pleural and pericardial effusions are common. Peripheral blood involvement is seen in approximately one-third of patients at time of presentation, and sometime during the clinical course in 80 % of patients with T-LBL who die from disease. Compared with patients with T-ALL, patients with T-LBL have lower serum LDH levels, a lower frequency of hepatosplenomegaly, and a lesser risk of central nervous system involvement [284]. T-LBL grows rapidly and, unless effectively treated, patients have a rapidly progressive downhill course with widespread dissemination of disease. Patients with T-LBL respond favorably to therapeutic regimens designed after those used for ALL.
Histologically, T-LBL replaces the lymph node in a diffuse pattern and often spills through the capsule into perinodal tissues [283, 285]. In cases with partial involvement, lymphoid follicles may be spared. A starry-sky pattern is present in a subset of cases. Rarely, the neoplasm is compartmentalized by fibrous tissue imparting a nodular low power pattern [286]. Cytologically, T-LBL is composed of lymphoblasts that have “dusty” or granular chromatin and either round or convoluted nuclear contours. Mitoses are numerous.
Immunophenotypic studies have shown that T-LBLs appear to illustrate the concept that these neoplasms represent clonal expansions of cells “frozen” in a state of differentiation [283, 284]. This phenomenon allows one to deduce that a variety of antigens are acquired in sequence, most likely reflecting the sequence of expression by nonneoplastic lymphoid cells during development. These stages of T-cell differentiation can be divided into pro-T, pre-T, cortical T, and medullary T [286, 287]. CD7 and cytoplasmic CD3 expression occur very early in this sequence and are expressed in the pro-T stage. The pro-T and pre-T stages are CD4−, CD8− and the cortical T stage is CD4+, CD8+, and CD1a+. TdT is expressed in all of these stages except the medullary T stage. T-LBLs also commonly express CD34, CD99, and BCL-2. The myeloid-associated antigens CD13 and CD33 can be expressed in up to one-third of cases [287, 288]. T lymphoblastic lymphomas are negative for Ig and pan-B-cell antigens, with the exception of CD79A which can be positive in a subset of T-LBLs, approximately 50 % in one study [289].
Cases of T-LBL and T-ALL (mostly T-ALL) have shown various patterns of T-cell receptor (TCR) gene rearrangement, corresponding to the stage of differentiation of the neoplasm [290, 291]. In a small subset of cases of T-ALL/LBL with a pro-T immunophenotype, only rearrangements of the TCRδ gene or the TCRδ and/or TCRγ genes may be identified. In the remaining tumors all of the TCR genes are rearranged. These findings suggest a hierarchy of TCR gene rearrangements in developing T lymphocytes: the TCRδ gene rearranges first, followed by the TCRγ and TCRβ genes, and lastly the TCRα gene. T-ALL/LBL cases also exhibit lineage infidelity manifested by the presence of IgH gene rearrangements in approximately 20 % of cases [283, 291].
Most cytogenetic and molecular studies performed on T-cell lymphoblastic neoplasms have been performed on cases of T-ALL, with relatively few cases of T-LBL assessed. Based on the available data, there is clearly overlap between T-LBL and T-ALL, but it must be noted that the cytogenetic and molecular profile for T-LBL presented here is based primarily on T-ALL data. Conventional cytogenetic studies of T-ALL/LBL have shown abnormalities in approximately 50 % of cases [284, 292]. The most common translocations involve the TCR genes at chromosomal loci 14q11 (TCRα and δ), 7q35 (TCRβ), and 7p14–15 (TCRγ). The most frequent gene partners are HOX11/TLX1 at 10q24 and HOX11L2/TLX3 at 5q35, with many other partners including TAL1 (1p32), RBTN1/LMO1 (11p15), RBTN2/LMO2 (11p13), LYL1 (19p13), and MYC (8q24). These translocations are of two types: (1) oncogenes are maintained intact but are juxtaposed with TCR gene loci; (2) two genes are disrupted creating a novel, chimeric gene. Many additional cytogenetic or molecular alterations have been shown in T-ALL, most of which affect the cell cycle. NOTCH1 gene mutations are common in T-ALL, in 40–50 % of cases, and additional abnormalities in other genes in the NOTCH1 pathway have been reported, such as loss of PTEN and AKT activation [293]. Additional somewhat common alterations in T-ALL include: deletions of chromosome 9p21 (30–40 %), del6q (in 15–20 %), WT1 mutations (∼10 %), duplications/gains of chromosome 6q23/MYB (5–10 %), rearrangements/amplification of ABL1 (5 %), protein tyrosine phosphatase 2 (PTPN2) mutations, and deletions of chromosome 13q14/RB1 [294–298].
Gene expression profiling of T-ALL cases has shown distinct gene signatures that correlate with the known cytogenetic abnormalities [299]. These studies also have shown that T-ALL cases without identified cytogenetic abnormalities can be characterized by these gene signatures suggesting the presence of cytogenetically undetectable molecular abnormalities in these cases. There are at least five gene signature groups that correlate, in part, with immunophenotype: HOX11/TLX1 (early cortical T), HOX11L2/TLX3 (early cortical T), TAL1 plus either LMO1 or LMO2 (late cortical T), LYL1 plus LMO2, and MLL-MLLT1 (pro-T). Comparisons of T-ALL and T-LBL have been few to date: T-LBL cases preferentially express genes encoding cell adhesion and extracellular matrix proteins [300].
Adult T-Cell Lymphoma/Leukemia
Adult T-cell lymphoma/leukemia (ATLL) is a type of mature T-cell lymphoma that is caused by infection by the human T-cell leukemia/lymphoma retrovirus
(HTLV-I) [301]. ATLL accounts for the high incidence of T-cell lymphoma in Japan, particularly on the islands of Kyushu and Okinawa, where up to 30 % of normal individuals are carriers with serologic evidence of HTLV-1 infection [302]. Other endemic regions for HLTV-I infection include Brazil and Caribbean countries and cases are also reported, albeit infrequently, in Europe and the United States [303]. HTLV-I is a single-stranded RNA retrovirus that is lymphotropic for T-lymphocytes [301, 304]. The virus is transmitted by sexual intercourse, breast milk, shared needles among intravenous drug users, and transfusion of blood products. The virus, however, is insufficient to cause lymphoma by itself and therefore other abnormalities must occur. This sequence of events most likely takes years as the incubation period for development of ATLL in HTLV-I individuals ranges from 20 to 40 years [304].
Patients with ATLL are usually adults, with a median age between 58 and 62 years and a male to female ratio ranging from 1.2–1.5 to 1 [301, 305]. Affected patients may present with one of four variants: acute, lymphomatous, chronic, and smoldering [301, 306]. Patients with acute ATLL, the most common form of the disease, have generalized lymphadenopathy, hepatosplenomegaly, skin lesions, peripheral blood involvement with high leukocyte counts, lytic bone lesions, and hypercalcemia. Hypercalcemia also may develop in the absence of bone lesions. The lungs and central nervous system are commonly involved. Patients with the lymphomatous form of ATLL present with prominent lymphadenopathy and tumors in other organs without hepatosplenomegaly or hypercalcemia. Skin lesions are common. Many patients with the acute form of ATLL, and a lesser number with lymphomatous ATLL, have evidence of immunodeficiency and opportunistic infections. The IPI predicts prognosis in patients with acute/lymphomatous ATLL [305]. These patients respond to chemotherapy, but remissions are short and overall survival is usually poor [307].
Patients who present with chronic variant ATLL have an absolute lymphocytosis and cytologically abnormal cells in the blood. Skin lesions, lymphadenopathy, and involvement of other viscera may occur and survival is usually greater than 2 years [301, 306]. Patients who have the smoldering from of ATLL have chronic disease for years, usually skin lesions with minimal peripheral blood involvement; the viscera are usually spared. Patients with the chronic and smoldering variants of ATLL do not usually require therapy but should be carefully followed as approximately 25 % of patients transform to acute or lymphomatous variant ATLL [301, 307].
The neoplastic cells in the peripheral blood smear in patients with acute or lymphomatous ATLL are characteristic [301]. The circulating neoplastic cells are medium sized with basophilic cytoplasm and markedly irregular, multilobulated nuclei, including cloverleaf shapes, also known as flower cells. In lymph nodes and viscera, ATLL diffusely replaces lymph nodes or extranodal sites. Cytologically, ATLL exhibits a spectrum of cell sizes, including small, medium-sized, large, anaplastic, and Reed–Sternberg-like cells [301, 308]. Some cases of ATLL can resemble classical Hodgkin’s lymphoma or angioimmunoblastic T-cell lymphoma, but most cases are indistinguishable from cases of peripheral T-cell lymphoma, NOS without knowledge of HTLV-I infection.
Immunophenotypic studies have shown that ATLLs have a mature T-cell immunophenotype [301]. The neoplastic cells express the pan-T-cell antigens CD2, CD3, CD5, and the TCR αβ receptor, but are usually negative for CD7. Most cases of ATLL are CD4+ CD8−. The lymphoma cells intensely express CD25, and approximately 40 % of ATLLs are positive for CCR4 and FOXP3: markers of regulatory T-cells [309]. ATLL cells variably express CD30 in a subset of cases and are negative for Ig and B-cell antigens.
Conventional cytogenetic studies have shown a wide range of abnormalities in cases of ATLL, but without a consistent abnormality. Usually multiple abnormalities are identified per individual neoplasm, at least six in one study [310]. These results suggest a multistep pathogenesis for ATLL and are consistent with the known long incubation period from time of infection to onset of tumor.
The hallmark for the diagnosis of ATLL is the demonstration of HTLV-1 infection. Molecular identification of HTLV-I is ideal for this purpose as the virus clonally integrates into the host cell genome in a random fashion [304]. The site of HTLV-I integration into the genome appears to be similar in most cases; unusual sites of viral integration may correlate with a poorer prognosis [311]. The mechanisms to explain how HTLV-I is involved in lymphomagenesis are not well worked out. The TAX viral protein and HLTV-I basic leucine zipper factor (HBZ) are thought to be involved [301]. Another point worth mentioning is that the use of serum testing for HTLV-I infection as a surrogate for molecular evidence of HLTV-I infection is imperfect. In countries with a low prevalence of HLTV-I infection, the presence of HLTV-I antibody highly correlates with ATLL in a patient presenting with evidence of lymphoma. However, in Japan and other areas with a high prevalence of HTLV-I infection, antibody testing yields a high false positive rate [312].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


