Sex cord-stromal tumors
Pure stromal tumors
Fibroma
Cellular fibroma
Thecoma
Luteinized thecoma associated with sclerosing peritonitis
Fibrosarcoma
Sclerosing stromal tumor
Signet-ring stromal tumor
Microcystic stromal tumor
Leydig cell tumor
Steroid cell tumor
Steroid cell tumor, malignant
Pure sex cord tumors
Adult granulosa cell tumor
Juvenile granuloma cell tumor
Sertoli cell tumor
Sex cord tumor with annular tubules
Mixed sex cord-stromal tumors
Sertoli-Leydig cell tumors
Well differentiated
Moderately differentiated
With heterologous elements
Poorly differentiated
With heterologous elements
Retiform
With heterologous elements
Sex cord-stromal tumors, not otherwise specified
6.2 Sex Cord-Stromal Tumors: Pure Stromal Tumors
6.2.1 Fibroma, Cellular Fibroma, and Fibrosarcoma
6.2.1.1 Clinical Features
Fibroma is a benign tumor composed of fibroblasts and collagen fibers. The mean age of patients with ovarian fibroma is about 50 years. Cellular fibroma can recur, and so clinical follow-up is necessary. Fibrosarcoma is a malignant mesenchymal tumor with a poor prognosis [3]. The standard treatment involves complete resection followed by chemotherapy.
6.2.1.2 Pathological Features
Fibroma is firm with a smooth, lobulated surface and average size is 6 cm. Cellular fibroma is mainly composed of solid components with white cut surface. Fibrosarcomas are large and soft and typically exhibit necrosis and hemorrhaging. Microscopically, fibromas are composed of fusiform and uniform cells arranged in a fascicular or whorled pattern. The stroma is fibrous with focal hyalinization or calcifications; however, approximately 10% of fibromas are hypercellular (little collagenous stroma is seen). Cellular fibroma is defined as fibroma group tumor with high cellularity, mild to moderate nuclear atypia, and 3 or few mitotic figures in 10/10HPF(high power fields). Cellular fibromas may have mitotic activity of > 4/10HPF (mitotically active cellular fibroma) [4] (Fig 6.1). Many ovarian tumors that have been reported as fibrosarcomas would now be considered to be mitotically active cellular fibromas. Fibrosarcomas are characterized by cellular spindle cell fibromatous lesions with moderate to marked nuclear atypia, 4 or more mitotic figures per 10/HPF, and atypical mitotic figures and necrosis [3] (Fig. 6.2). Fibrosarcomas are usually large and have often spread beyond the ovary at diagnosis. Their differential diagnoses include leiomyosarcoma, high-grade endometrial stromal sarcoma, gastrointestinal stromal sarcoma, and various types of primary or metastatic soft tissue sarcoma.
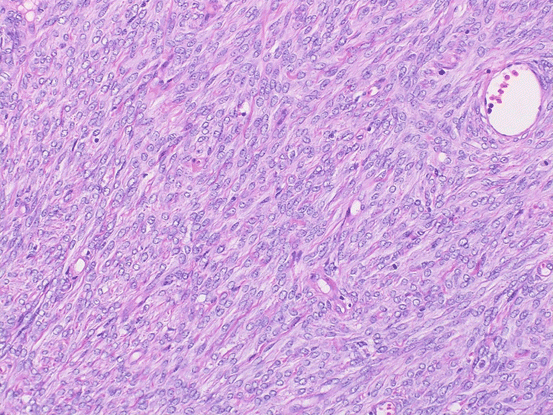
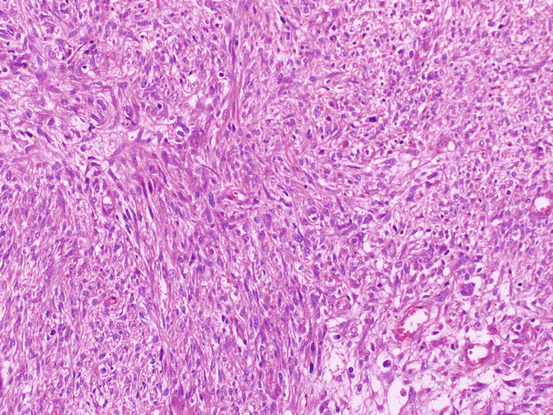
Fig. 6.1
Mitotically active cellular fibroma. Proliferating bland spindle-shaped cells and scattered mitotic figures are shown
Fig. 6.2
Fibrosarcoma. The fascicular proliferation of atypical spindle cells and bizarre giant cells is shown
6.2.2 Thecoma
6.2.2.1 Clinical Features
Thecomas are gonadal stromal tumors that are predominantly composed of theca cell-like cells. In daily practice, thecomas are uncommon, whereas fibromas are relatively common. Thecomas usually occur in premenopausal or postmenopausal women, but can arise in children in rare cases. Luteinized thecomas occur at a younger age, usually in patients in their 20s or 30s. Premenopausal women display either endocrine-associated symptoms, such as irregular bleeding or amenorrhea, or nonspecific complaints, such as pelvic pain or abdominal distention. Luteinized thecomas can be estrogenic (50%), androgenic (11%), or nonfunctional (39%) [5]. Some patients with luteinized thecomas are virilized, whereas others show hyperestrogenic symptoms. Thecomas are benign, and excision is an appropriate treatment. The diagnosis of luteinized thecoma is restricted to luteinized thecomas associated with sclerosing peritonitis, a distinctive stromal tumor that is typically associated with sclerosing peritonitis [1].
6.2.2.2 Pathological Features
Macroscopically, thecomas are firm or hard tumors with a mean diameter of 7 cm. The cut surfaces of thecomas are solid and yellow or white. Cysts and calcifications may be present.
Histologically, thecomas are composed of fascicles or sheets of plump spindle-shaped or ovoid stromal cells that resemble the cells of the theca interna. Tumor cells have round or fusiform nuclei and amphophilic or lightly eosinophilic or clear cytoplasm (Fig. 6.3). Mitotic figures are rare. A variable number of fibroblasts are intermixed among the theca-like cells. From a practical point of view, the diagnosis of thecoma is restricted to tumors that show evidence of steroid hormone secretion, have a conspicuous tumor composed of cells with clear or vacuolated cytoplasm, or contain luteinized cells (Fig. 6.4). Immunohistochemically, most thecomas express inhibin and calretinin.
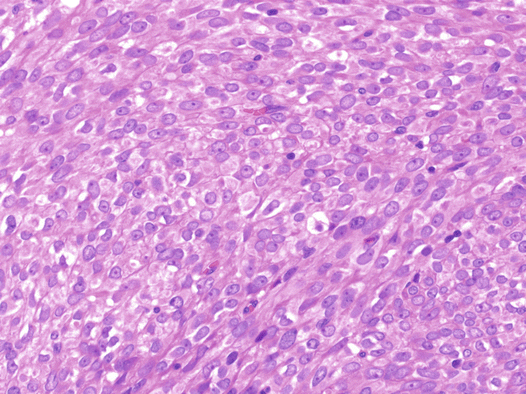
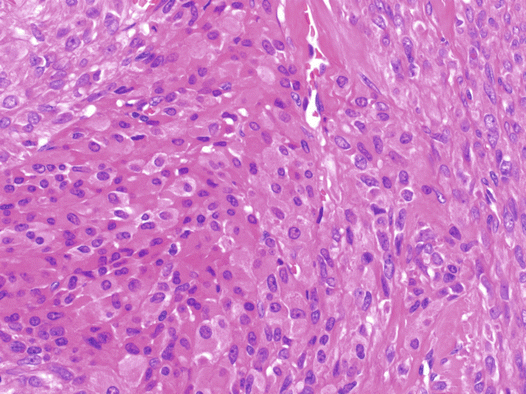
Fig. 6.3
Thecoma. Solid nests of cells with uniform round nuclei and lightly eosinophilic or clear cytoplasm are shown
Fig. 6.4
Luteinized thecoma. Sheets of luteinized cells with abundant eosinophilic cytoplasm are shown
6.2.3 Sclerosing Stromal Tumor
Sclerosing stromal tumors are uncommon benign stromal tumors that mainly occur in teenagers and young women [6]. They should be treated by excision or unilateral salpingo-oophorectomy. Macroscopically, these tumors are firm and white to yellowish-white. Histologically, they are characterized by the lobular proliferation of tumor cells with staghorn or hemangiopericytomatous vascular spaces. Tumor cells include polygonal theca-like cells with vacuolated eosinophilic cytoplasm and fibroblast-like cells (Fig. 6.5). Immunohistochemically, their cells are positive for vimentin, inhibin, calretinin, signal transducer activator of transcription 6 (STAT6), and the estrogen and progesterone receptors.
Fig. 6.5
Sclerosing stromal tumor. The tumor is composed of polygonal theca-like cells with vacuolated eosinophilic cytoplasm, fibroblast-like cells, and staghorn vascular spaces
6.2.4 Microcytic Stromal Tumor
6.2.4.1 Clinical Features
This tumor has recently been described by Irving and Young [1] as a previously uncharacterized ovarian neoplasm that exhibits prominent microcystic changes and is most likely of stromal origin. The reported cases involved patients who ranged in age from 26 to 63 (mean, 45) years, and most patients presented with a pelvic mass. Hormonal manifestations are rarely seen. Microcytic stromal tumors are unilateral and do not undergo extraovarian spread.
6.2.4.2 Pathological Features
Microcystic stromal tumors are solid-cystic, solid, or predominantly cystic and display a mean diameter of 8.7 cm. Their solid components are firm and tan or white-tan. Microscopically, these ovarian tumors contain microcysts with variable amounts of solid cellular tissue and fibrous stroma. In addition, they exhibit lobular demarcation as well as sharp separation from the ovarian stroma. The microcysts are characterized by small round to oval cystic spaces. Intracytoplasmic lumens or vacuoles are also present (Fig. 6.6). The tumor cells contain moderate abundant finely granular, eosinophilic cytoplasm and bland, round to oval or spindle-shaped nuclei with fine chromatin and indistinct nucleoli [7]. Mitotic figures are very rare. This type of tumor is characterized by an absence of morphological features that would result in any other specific diagnosis in the sex cord-stroma category, an absence of epithelial elements, and an absence of teratomatous or other germ cell elements [7]. Immunohistochemically, the tumors are strongly positive for CD10 and vimentin, but do not express S-100 protein, calretinin, inhibin, epithelial membrane antigen (EMA), cytokeratin (CK), melan A, and estrogen receptors, desmin, chromogranin A, synaptophysin, WT1, or CD34 [1].
Fig. 6.6
Microcystic stromal tumor. The microcysts are characterized by small round to oval cystic spaces. Intracytoplasmic lumens or vacuoles are also present
6.3 Pure Sex Cord Tumors
6.3.1 Adult Granulosa Cell Tumor (AGCT)
6.3.1.1 Clinical Features
Granulosa cell tumor is the most common type of malignant sex cord-stromal tumor. There are two types of granulosa cell tumor, the adult type, which mainly occurs in premenopausal and postmenopausal women (mean age, 45–55 years), and juvenile granulosa cell tumors (JGCT), which mainly occur in children (mean age, 15 years) [1]. It is important that the distinction between AGCT and JGCT is made on the basis of the histology and not the patient age. The typical clinical presentation of AGCT is postmenopausal bleeding in older women and menorrhagia or amenorrhea in younger patients. Granulosa cell tumors typically secrete estrogen, and patients with these tumors exhibit endometrial hyperplasia (30–40%) or adenocarcinoma (5–10%) [8]. Granulosa cell tumors are typically unilateral and confined to the ovary at diagnosis.
The overall recurrence rate ranges from 10 to 30%. Metastases or recurrence is often detected more than 5 years after the initial treatment, particularly in the peritoneum and omentum. There is no correlation between the microscopic features of tumors, including mitotic activity, and outcomes.
6.3.1.2 Pathological Features
Macroscopically, most AGCT are solid and cystic. The solid areas are soft to firm and yellow/brown to tan. Some tumors are predominantly cystic. The average size is about 10 cm. A variety of growth patterns are observed in AGCT, including admixtures of different patterns. The cells of such tumors often grow in microfollicular or diffuse patterns. Granulosa cell tumors consist of nests and sheets of granulosa cell-like cells punctuated by small spaces, which resemble Call-Exner bodies (Fig. 6.7). Occasionally, larger follicles are sometimes observed (the macrofollicular pattern). The cells of granulosa cell tumors are often arranged in cords, trabeculae, and ribbons (Fig. 6.8). In addition, they have scant pale cytoplasm and uniform, pale, round oval nuclei. Coffee bean-like nuclear grooves were considered to be a characteristic of granulosa cell tumors, but they are not seen every case, and they also occur in many other neoplasms, including Sertoli cell tumors and Sertoli-Leydig cell tumors. Brisk mitotic figures are seen in some lesions. Some AGCT have a JGCT component, and such tumors should be classified based on their predominant histology.
Fig. 6.7
Adult granulosa cell tumor. The tumor cells are uniform and have grooved nuclei. Note the numerous Call-Exner bodies
Fig. 6.8
Adult granulosa cell tumor. The tumor cells grow in cords, trabeculae, and ribbons
Immunohistochemically, granulosa cell tumors are usually positive for inhibin, calretinin, FOXL2 (forkhead box L2), WT1, and CD56, whereas they are negative for CK7 and EMA. A missense somatic point mutation that is characteristic of AGCT has recently been identified in the FOXL2 gene [9]. This mutation is seen in approximately 95% of AGCT.
6.3.2 Juvenile Granulosa Cell Tumor (JGCT)
6.3.2.1 Clinical Features
Fewer than 5% of granulosa cell tumors occur in children or teenagers. Almost all JGCT are unilateral, and more than 95% of them are confined to the ovary (stage I). The symptoms of JGCT are often caused by the estrogen they secrete. Young girls frequently display isosexual pseudoprecocity, whereas older children and premenopausal women develop menstrual abnormalities or amenorrhea. Associations have been detected between JGCT and Ollier (enchondromatosis) disease and Maffucci (enchondromatosis and multiple hemangiomas) syndrome [10]. The prognosis of patients with JGCT is better than that of patients with AGCT. JGCT are less likely to recur or metastasize. The long-term survival of patients with JGCT is good, but patients whose tumors rupture or who exhibit positive peritoneal cytology or extraovarian tumor spread are at significant risk of recurrence. Inhibin and Müllerian inhibitory substance are useful tumor markers for following up patients with JGCT.
6.3.2.2 Pathological Features
The average size is about 12 cm, and most of them exhibit a mixed solid-cystic appearance, but some are completely solid or cystic. Their solid areas are yellow or tan. Hemorrhaging is sometimes seen, but necrosis is uncommon.
Microscopically, JGCT show a multinodular growth pattern, and macrofollicular, solid, and cystic growth patterns are characteristic of JGCT. Follicles often vary in size and shape and contain mucinous material (Fig. 6.9), macrofollicles are lined by one or more layers of granulosa cells and are surrounded by a rim of theca cells. Solid areas are composed of sheets of granular cells made up of an admixture of theca cells or fibroblasts. The microfollicular, insular patterns and trabeculae seen in AGCT are rarely observed in JGCT. Tumor cells have large and round nuclei and amphophilic or pink cytoplasm. In addition, they lack coffee bean-like nuclear grooves and may contain conspicuous nucleoli. Some tumor cells have enlarged pleomorphic nuclei, and multinucleated cells can also be observed (Fig. 6.10). Mitotic figures tend to be numerus with an average around 6/10HPF.
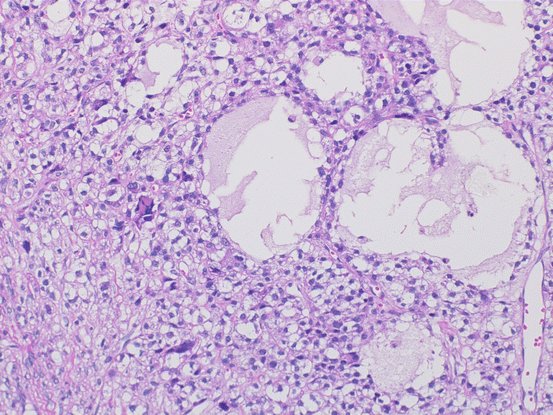
Fig. 6.9
 Ovarian Cancer Genome and Molecular Experimental Sciences
Strategies for the Management of Epithelial Ovarian Cancer
Ovarian Cancer Genome and Molecular Experimental Sciences
Strategies for the Management of Epithelial Ovarian Cancer
 Management of Ovarian Cancer in the Elderly Population
Management of Ovarian Cancer in the Elderly Population
 Strategies for the Management of Non-epithelial Ovarian Tumors
Strategies for the Management of Non-epithelial Ovarian Tumors
 Strategies for the Management of Epithelial Ovarian Borderline Tumors
Strategies for the Management of Epithelial Ovarian Borderline Tumors
 Pathology of Epithelial Ovarian Tumors
Pathology of Epithelial Ovarian Tumors




Juvenile granulosa cell tumor. The tumor shows macrofollicular, solid, and cystic growth patterns. The follicles vary in size and shape
Related posts:
Ovarian Cancer Genome and Molecular Experimental Sciences
Strategies for the Management of Epithelial Ovarian Cancer
Management of Ovarian Cancer in the Elderly Population
Strategies for the Management of Non-epithelial Ovarian Tumors
Strategies for the Management of Epithelial Ovarian Borderline Tumors
Pathology of Epithelial Ovarian Tumors
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
