Germ cell tumours
Dysgerminoma
Variant
With syncytiotrophoblastic cells
Yolk sac tumours
Variants
Polyvesicular vitelline
Hepatoid
Glandular
Embryonal carcinoma
Polyembryoma
Choriocarcinoma, non-gestational
Teratoma
Immature
Mature
Solid
Cystic (dermoid cyst):
With secondary tumour (specify type)
Fetiform
Monodermal
Struma ovarii
With secondary tumour (specify type)
Carcinoid
Insular
Trabecular
Strumal carcinoid
Mucinous carcinoid
Neuroectodermal tumour (specify type)
Sebaceous tumour
Mixed germ cell tumour (specify types)
Sex cord-stromal tumours
Granulosa-stromal cell tumours
Granulosa cell tumour
Adult
Juvenile
Tumours in thecoma-fibroma group
Thecoma
Typical
Luteinised
Malignant
Fibrothecoma
Fibroma
Typical
Cellular
With sex cord elements
Fibrosarcoma
Sclerosing stromal tumour
Sertoli–stromal cell tumours
Well differentiated
Sertoli cell tumour
Sertoli–Leydig cell tumour
Sertoli–Leydig cell tumour of intermediate differentiation
Variant: With heterologous elements
Sertoli–Leydig cell tumour, poorly differentiated
Variant: With heterologous elements
Retiform
Variant: With heterologous elements
Sex cord tumour with annular tubules
Gynandroblastoma
Steroid cell tumours
Leydig cell tumour, hilus and stromal variants
Adrenal rest tumour
Steroid cell tumour, not otherwise specified
Mixed germ cell and sex cord-stromal tumours
Gonadoblastoma
Variant: With dysgerminoma or other germ cell tumour
Germ cell sex cord tumour on non-gonadoblastoma type
Variant: With dysgerminoma or other germ cell tumour
Tumours of rete ovarii
Adenoma and cystadenoma
Adenocarcinoma
Mesothelial tumours
Adenomatoid tumour
Mesothelioma
Tumours of uncertain origin and miscellaneous tumours
Small cell carcinomas
Tumour of probable Wolffian origin
Hepatoid carcinoma
Myxoma
Gestational trophoblastic diseases
Soft tissue tumours not specific to ovary
Malignant lymphomas and leukaemia
Germ Cell Tumours
Germ cell tumours are a group of gonadal, and rarely extragonadal, neoplasms with identical counterparts in the ovary and the testis, albeit with biological and clinical diversity [2, 3]. Ovarian germ cell tumours are classified by the WHO into primitive germ cell tumours and teratomas [4]. The primitive germ cell tumours are dysgerminoma, yolk sac tumour, embryonal carcinoma and choriocarcinoma. The prognostic significance of each tumour type has altered significantly after the introduction of combination chemotherapy, improving survival rates to almost comparable levels [5–7]. Teratomas are neoplasms containing components from the three germ cell layers (endoderm, mesoderm and ectoderm) and can contain derivatives from one, two or all three layers. Hence, they are subclassified as biphasic/triphasic teratomas, the most common being the dermoid cyst, and monodermal teratomas, represented by struma ovarii and ovarian carcinoid. Biphasic and triphasic teratomas are further categorised as mature or immature teratomas, a distinction based on the presence of immature neuroepithelium. Mature teratomas are benign while immature teratomas behave in a malignant fashion.
Germ cell tumours constitute the second largest group of ovarian tumours after surface epithelial tumours and comprise 30 % of all ovarian neoplasms. The vast majority (95 %) are benign mature cystic teratomas (dermoid cysts) [8]. Malignant germ cell tumours are rare, representing 3–5 % of ovarian cancers in Europe and America, while in Asia and Africa, and especially in Japanese women, where surface epithelial tumours are less common, their relative incidence is higher, up to 20 % [9, 10]. Malignant germ cell tumours are almost always encountered in children, adolescents and young women, in which group of patients maintenance of fertility is an important clinical issue. In this age group, they represent the most common ovarian cancer, with 60 % of ovarian tumours up to the age of 21 being germ cell tumours, and one-third of them being malignant [11]. Overall, malignant germ cell tumours are rare beyond the fourth decade of life. In older adults, the majority of germ cell neoplasms are mature cystic teratomas, although case reports of primitive germ cell tumours in postmenopausal women have been published where they appear to arise from surface epithelial tumours [12].
Germ cell tumours originate from the primitive germ cells, which migrate to the gonadal anlage from the yolk sac endoderm. They reach the genital ridge during the fifth and sixth week of embryonic life. During embryogenesis, these primordial germ cells will ultimately form the primary oocytes through developmental stages of mitosis, atresia and meiosis. Different germ cell tumours are considered to arise from germ cells at different stages of development in the ovarian gonad. A widely held hypothetical model of histogenesis by Teilum regards seminoma (and thereby dysgerminoma) as immediately arising from germ cells without any further differentiation [13–15]. Embryonal carcinoma, on the other hand, is regarded to arise from germ cells with pluripotential dynamics of differentiation, and in this context ‘embryonal carcinoma’ has a dual meaning: a conceptual term of tumourigenesis and an actual tumour type. According to Telium’s model, tumourigenesis can then follow two pathways. One is the embryonic pathway, producing endoderm, ectoderm and mesoderm, thereby producing teratomas. The second follows the extraembryonic pathway of differentiation towards endodermal sinus tumour (yolk sac tumour) and choriocarcinoma (tumour recapitulating trophoblast). This dynamic process of differentiation explains why some of these components can coexist in mixed germ cell tumours [16].
Germ cell tumours usually present with abdominal pain, most likely from ovarian torsion, the presence of a mass and/or abdominal distension [17, 18]. Although they are hormonally inert tumours, unusual presentations include pregnancy-like presentation due to human chorionic gonadotropin (HCG) secretion and hyperandrogenaemia [19]. Very rarely, germ cell tumours present during pregnancy, raising management issues of treatment and pregnancy salvation [20]. Tumour markers such as alpha-fetoprotein (AFP) and beta-HCG (bHCG), produced by the tumour cells, are useful clinical tools for diagnosis and follow-up after treatment. Radiological diagnostic differentiation of germ cell tumour types is difficult, with the exception of computed tomography (CT) identification of Rokitansky protuberances and fatty cyst contents in mature cystic teratomas.
Dysgerminoma
Dysgerminoma, the ovarian counterpart of testicular seminoma, is the most common malignant germ cell tumour of the ovaries, accounting for 3–5 % of ovarian cancer and 1–2 % of primary ovarian neoplasia [21, 22]. The median age at diagnosis is 22 years, although it has been reported in infants and in women of up to 70 years of age [23, 24]. The clinical symptoms are similar to those of most ovarian neoplasms, i.e. abdominal pain due to ovarian torsion, presence of a mass and abdominal distension. It is a rapidly growing mass presenting as a large tumour within a short period of time. Dysgerminomas with syncytiotrophoblasts or with luteinised stromal cells may present with endocrine symptomatology [25]. Rare cases present with hypercalcaemia due to parathormone-related peptide or 1,25-dihydroxyvitamin D secretion [26, 27]. On imaging, the ultrasound finding of a highly vascularised, large, solid, lobulated adnexal mass with irregular internal echogenicity, in a woman 20–30 years old, raises the suspicion of ovarian dysgerminoma [25]. It is important to note that the traditional tumour markers for germ cell tumours are not raised: AFP is never elevated except in cases with an additional germ cell component (i.e. as a mixed germ cell tumour) and HCG only in 3–5 % of cases [28]. Serum lactate dehydrogenase is usually raised and useful for monitoring treatment and recurrence [29].
Dysgerminoma is most commonly unilateral but it may also be bilateral, with gross or microscopic evidence of contralateral ovarian involvement. It is a well-circumscribed, encapsulated tumour with a solid, uniform, lobular cut surface, approximately 15 cm in size, although larger masses up to 50 cm have been recorded in the literature [30]. They may show areas of necrosis, cystic change and calcification. When the tumours are bilateral and calcification is sandy and pinpoint, the presence of concurrent gonadoblastoma may be considered. Cystic change suggests the presence of a second germ cell tumour component, especially a teratoma. The recommended sampling of ovarian tumours dictates one section per centimetre of maximum dimension, with additional sampling of heterogenous areas, to exclude the presence of other germ cell tumour components.
The typical histological appearances of dysgerminoma (Fig. 5.1) resemble those of seminoma. The architectural pattern is that of a nodular, lobulated tumour or of tumour cells arranged in islands, cords and strands, depending on the amount of stroma present, which may vary from scanty to abundant. The tumour cells are large cells with distinct cell membranes, high nuclear cytoplasmic ratio (almost 50 %), showing round nuclei with a prominent nuclear membrane, uneven granular chromatin and one or two nucleoli. Mitotic activity may be scanty or brisk and does not correlate with behaviour [31]. The cytoplasm is slightly granular and eosinophilic to clear and contains glycogen and fat. The stroma is composed of fibrous tissue forming septa separating the tumour into lobules, to hyalinised or oedematous areas or abundant dense fibrous tissue, causing variable cellularity within the same tumour and between tumours. It characteristically contains a lymphocytic infiltrate, which is a diagnostic pointer towards dysgerminoma, and although it is mostly composed of T lymphocytes, it may also include follicles with germinal centres. Foreign body giant cell reaction and granuloma formation may also be seen within the stroma. The tumours contain thin-walled vessels within the stroma. Syncytiotrophoblastic cells form an inconspicuous to prominent component of the tumour. Their presence is associated with raised levels of bHCG in the serum, but at levels much lower than those seen in choriocarcinoma, which can be used for tumour monitoring. Syncytiotrophoblastic cells are sometimes seen in close proximity to areas of haemorrhage within the tumour, and care should be taken to exclude the presence of choriocarcinoma by making sure there is no cytotrophoblast component intermixed with the syncytiotrophoblastic cells, which is the diagnostic hallmark of choriocarcinoma. Areas of haemorrhage with or without syncytiotrophoblast can be encountered in pure dysgerminomas without any choriocarcinomatous component. Also, luteinised stromal cells can rarely be seen and their presence may be related to symptoms of virilisation or feminisation.
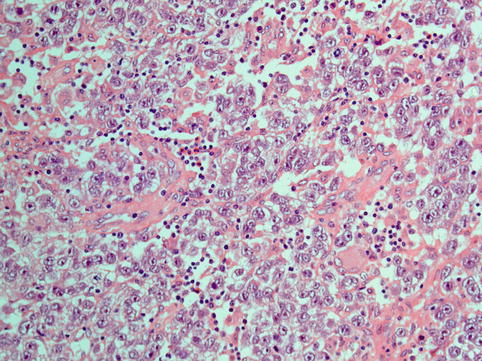
Fig. 5.1
Dysgerminoma is composed of solid sheets of cells with abundant clear to pale cytoplasm and atypical vesicular nucleoli with prominent nucleoli. Infiltration of fibrous septa by mature lymphocytes is a characteristic feature
In similarity to their testicular counterparts, molecular analysis in dysgerminomas has shown chromosome 12p abnormalities, including i12p and 12p over-representation. Fluorescence in situ hybridisation (FISH) analysis for chromosome 12p abnormalities may be a useful diagnostic adjunct for confirming the diagnosis of dysgerminoma, but i12p has also been reported in non-dysgerminomatous germ cell tumours [32, 33]. C-KIT mutations have also been identified in ovarian dysgerminomas similar to testicular seminomas. They tend to occur in advanced stage tumours and may pose a target for therapeutic treatment for those dysgerminomas that carry the mutation [21].
Dysgerminoma is positive for seminoma markers placental alkaline phosphatase (PLAP) (membranous), OCT3/4 (nuclear), CD117, SALL4 and D2-40. It should be noted that occasional cells are positive for low molecular weight keratins (e.g. CAM 5.2). PLAP is positive in all malignant germ cell tumours and its use does not offer any differential diagnostic value between germ cell tumours, but it does aid in the differential diagnosis from non-germ cell tumours (lymphoma, carcinoma). OCT3/4 is positive in dysgerminomas and embryonal carcinomas only, and therefore offers some specificity. CD117 is positive in nearly all dysgerminomas, although this does not indicate an underlying mutation which is only seen in a minority of cases [21, 34]. SALL4 is expressed in primitive germ cell tumours and it is also encountered in gastric carcinomas with hepatoid differentiation [35]. D2-40 is not specific for dysgerminoma as it is expressed in a variety of human tumours; however, dysgerminoma is the only germ cell tumour in which D2-40 is expressed. Low molecular weight keratin helps in the differential diagnosis from embryonal carcinoma and yolk sac tumour, which are both diffusely positive, in comparison with occasional cell positivity in dysgerminoma.
Dysgerminoma must be differentiated from lymphoma, clear cell carcinoma, granulosa cell tumour, embryonal carcinoma, yolk sac tumour and steroid cell tumours. Characteristic features of dysgerminoma are the nodularity of the tumour, tumour cells with more or less uniform cytological atypia and closely intermixed lymphoid infiltrate, as well as immunohistochemical expression of PLAP, OCT3/4 and CD117. Clear cell carcinoma, embryonal carcinoma and yolk sac tumour, even if predominantly solid, show areas of papillary and glandular architecture which can be identified as minor components following careful examination and thorough sampling. Also, embryonal carcinoma shows greater nuclear pleomorphism. Steroid cell tumours are encountered in older patients than dysgerminomas and they show finely vacuolated cytoplasm with positive staining for inhibin and calretinin.
Dysgerminoma is a rapidly growing tumour with metastatic potential, more often through the lymphatic than the haematogenous route, but with less aggressive behaviour than the rest of the malignant germ cell tumours. This tumour responds well to chemotherapy, even in the presence of disseminated disease, as well as radiotherapy. Its prognosis is excellent for stage I and stage III retroperitoneal disease with worsening outcome in the presence of peritoneal spread [22, 36]. When disease is confined to one ovary without rupture, treatment may consist of unilateral oophorectomy or salpingo-oophorectomy alone for preservation of fertility. The role of adjuvant chemotherapy in these cases is controversial [7, 30, 31, 37].
Yolk Sac Tumour
Yolk sac tumours constitute a multifaceted group of malignant neoplasms for which the term primitive endodermal tumours may be more suitable, as they differentiate towards various extraembryonal and somatic directions. The endodermal sinus is actually an exceptional pattern of differentiation in these tumours but the name has prevailed. During embryogenesis, yolk sac first develops in its primary form made of primitive endoderm cells which form the lining epithelium of the sac, while on day 13 it evolves to a secondary embryonal sac composed of a combination of primitive endodermal cells and mesoblast. In the 13th week, the primitive endoderm is replaced by the embryonic endoderm. From this the three somatic germ layers will emerge. This embryological development may explain the diversity in the morphological patterns encountered in these tumours [38].
Of the ovarian primitive germ cell tumours, yolk sac tumours are second in frequency after dysgerminomas. These occur in children and young adults while exceedingly rare case reports of presentation in postmenopausal women exist [12]. They present with similar symptoms to other germ cell tumours. These can present in pregnancy but are not associated with endocrine manifestations as are some other types of malignant germ cell tumours. High levels of serum AFP are almost pathognomonic of the tumour, although raised levels of AFP at a lower concentration are seen in other tumours, such as hepatoid carcinoma, embryonal carcinoma and Sertoli–Leydig tumours [39–41].
Yolk sac tumours are commonly unilateral. If there is a concurrent tumour in the contralateral ovary it is often a mature teratoma [42] and bilateral yolk sac tumours signify metastatic disease. Yolk sac tumours are usually encapsulated, large tumours which have a characteristic grey yellow micro- or macrocystic cut surface or may show areas of necrosis, haemorrhage and liquefaction.
Histologically there is a diversity of architectural patterns encountered in these tumours which can make tumour typing rather difficult. The characteristic reticular pattern should be sought, and its presence makes the diagnosis easier. The reticular/microcystic pattern is composed of a loose myxoid stroma associated with lacunar spaces lined by attenuated epithelium of variable degrees of atypia (Fig. 5.2) with or without periodic acid–Schiff (PAS)-positive hyaline globules, reminiscent of the mesodermal/endodermal tissue pattern encountered in yolk sac. Schiller–Duval bodies (Fig. 5.3) are papillaroid structures with a fibrovascular core lined by a layer of cuboidal to columnar cells showing vesicular nuclei, prominent nucleoli and eosinophilic to clear cytoplasm. These papillae project into spaces which are lined by a monolayer of cells with hyperchromatic nuclei. These structures have been identified in murine placentas and they recapitulate endodermal sinuses, constituting the hallmark of these tumours. Histological variants are polyvesicular vitelline, solid, parietal, hepatoid and glandular (e.g. intestinal, pulmonary).
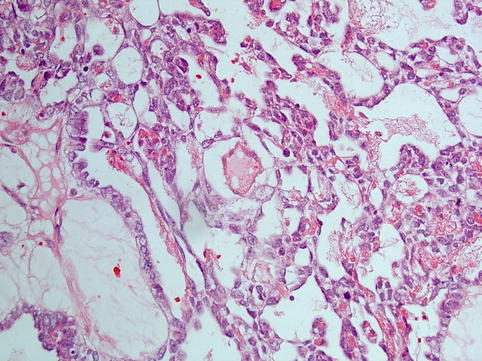
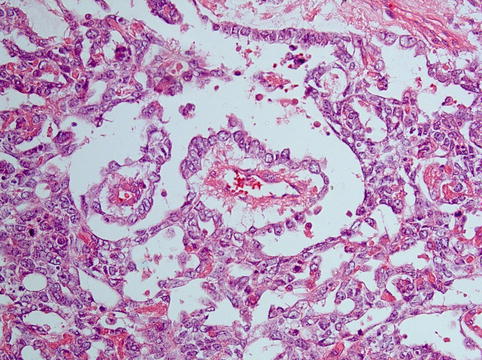
Fig. 5.2
Yolk sac tumour: tumour arranged in a reticular/microcystic pattern, composed of a loose myxoid stroma associated with lacunar spaces lined by attenuated epithelium showing variable atypia
Fig. 5.3
Schiller–Duval body in a yolk sac tumour, papillaroid structure with a fibrovascular core lined by a layer of cuboidal to columnar cells projecting into a space lined by a monolayer of flattened cells with hyperchromatic nuclei
On immunohistochemistry, yolk sac tumour cells are positive for AFP, glypican-3 and SALL4 [43–45]. They are positive for pancytokeratin markers but usually negative for CK7 and EMA [46]. Yolk sac tumours may be positive for PLAP but are negative for OCT3/4 and CD117 [2].
Yolk sac tumours usually have characteristic morphological features but may need to be differentiated from dysgerminomas, embryonal carcinomas, endometrioid carcinomas and clear cell carcinoma of the ovary and rarely from juvenile granulosa cell tumours. Typical features of dysgerminoma, such as a lymphocytic infiltrate closely intermixed with tumour cells and expression of CD117 and OCT3/4, aid in the differential. Solid forms of yolk sac tumour may be mistaken for embryonal carcinomas. The nuclear pleomorphism in the latter is greater, and even when it is better differentiated, it does not form the structures encountered in yolk sac tumour. Finally, yolk sac tumours are much more common than embryonal carcinomas as ovarian malignant germ cell tumours. The glandular variant of yolk sac tumour occasionally mimics endometrial secretory pattern with subnuclear vacuolation, in which case it needs to be differentiated from endometrioid ovarian adenocarcinoma. Oestrogen (ER) and progesterone (PR) positivity and squamous differentiation favour endometrioid adenocarcinoma. When clear cell carcinoma of the ovary is predominantly composed of a tubular architectural pattern, its tubules show a typical hobnail pattern of their epithelial lining and they are not lined partly by cuboidal and partly by flattened mesothelial-like epithelium, as is yolk sac tumour, especially the perivitelline variant. Inhibin helps in the differential diagnosis between yolk sac tumour and sex cord-stromal tumours such as granulosa cell tumour with macrofollicular pattern resembling yolk sac tumour and Sertoli–Leydig cell tumours with raised AFP.
With the advent of multiagent chemotherapy, an overall 80 % complete cure rate for all stages of this tumour has been achieved, transforming the previous poor prognosis of this highly malignant tumour, which metastasises to lymph nodes and organs and also spreads by direct extension [47].
Teratoma
Teratomas are broadly classified into mature and immature, immature teratomas being the malignant representatives of this group. They are the only tumours within the group of malignant germ cell tumours which are graded, as grading has prognostic significance and determines the use or not of adjuvant chemotherapy. Mature (cystic) teratomas (dermoid cysts) are the most common germ cell tumours of the ovary. These are benign but rarely may be associated with somatic transformations of one of their components into malignant tumours, the most common being squamous cell carcinoma, adenocarcinoma or adenosquamous carcinoma. Another feature of clinical significance regarding mature teratomas is the fact that they can be associated with autoimmune haemolytic anaemia [48]. This dictates pelvic and lower abdominal investigation for a mature teratoma in the workup of autoimmune haemolytic anaemia in a female child or young woman to avoid inappropriate splenectomy. Although very rare, solid mature teratomas may contain immature components which, if present, lead to tumour recurrence. Monophasic teratoma is a mature teratoma deriving from a single germ cell layer. The most common is struma ovarii, which consists entirely of normal or hyperplastic thyroid tissue. Rarely thyroid tissue in struma ovarii may show neoplastic change towards benign or malignant thyroid tumours such as follicular adenoma, follicular carcinoma and papillary carcinoma, with the latter being the most common neoplasm. Histogenetically, all teratomas appear to be the product of parthenogenesis, arising from a primordial germ cell after implantation in the ovarian gonad, post first meiotic division.
Immature Teratoma
Immature teratomas have an age range of presentation in the first two to three decades of life, being almost unheard of in postmenopausal women [49, 50]. They present as most other germ cell tumours and they grow much more rapidly than mature teratomas, although when the latter have reached a significant size at the time of diagnosis this feature is no longer helpful.
Immature teratomas are usually unilateral solid or less commonly solid and cystic masses, which are often associated with capsular rupture and peritoneal spread. They have a grey white to haemorrhagic and necrotic cut surface, while areas resembling cartilage or bone may be present.
Immature teratomas contain embryonal type and immature tissues of all three germ cell layers, endoderm, mesoderm and ectoderm. It is essentially the presence of immature neuroepithelium that is not only diagnostic but also of prognostic value in being the basis for grading. Other forms of immature tissue may be present but in the absence of immature neuroepithelium, or rarely immature glial tissue, are in themselves insufficient for a diagnosis of immature teratoma [2]. Less mature tissue may be present in mature teratomas as well, but in immature teratomas these are more disorganised with no organoid appearance. A grading system devised by AFIP and incorporated by WHO is applied in both the primary ovarian tumour and any peritoneal deposits (Tables 5.2 and 5.3 [50, 51]). Immature neuroepithelium is formed of small blue cells with formation of rosettes and brisk mitotic activity (Fig. 5.4) [50]. Immature mesoderm presents as loose myxoid stroma containing immature mesenchymal components such as immature cartilage and rhabdomyoblasts. Less common are immature endodermal structures, which may be glands with basal vacuolation or embryonic renal tissue resembling Wilms tumour.
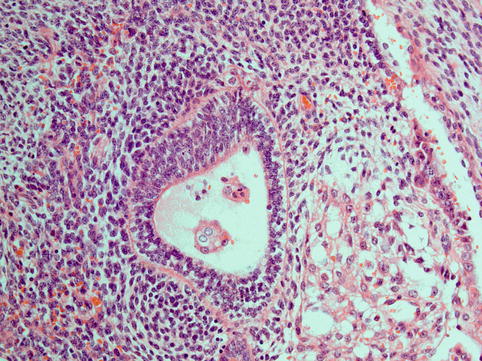
Grade 1 | Immature neuroepithelium <1 low power field (X4 objective) in one slide |
Grade 2 | Immature neuroepithelium 1–3 low power fields (X4 objective) in one slide |
Grade 3 | Immature neuroepithelium >3 low power fields (X4 objective) in one slide |
Three-tier grading | Combination chemotherapy |
|---|---|
Grade 1 ovarian tumour | Not required |
Grade 2 or 3 ovarian tumour | Required |
Grade 2 or 3 implants | Required |
Grade 0 implants regardless of ovarian tumour grade | Not required |
Fig. 5.4
Immature teratoma is characterised by variable amounts of immature neuroepithelium and neuroblastoma-like areas
Immunohistochemistry does not have such a useful place in the diagnosis of immature teratoma and the diagnosis is essentially morphological, although some markers have recently been suggested to have prognostic value [52]. Immature teratomas should be differentiated from carcinosarcomas or malignant mixed Müllerian tumours of the ovary, also tumours with a mixture of epithelial and mesenchymal components which occur in older women rather than the younger target group of immature teratomas. The presence of adenocarcinoma and malignant stromal elements versus the immature elements of teratoma aids the differential diagnosis.
Unilateral salpingo-oophorectomy with or without triple chemotherapy is the treatment of choice. The use of chemotherapy is determined by grading and the presence of metastatic disease. Immature teratomas may metastasise to lymph nodes and other organs and the phenomenon of chemotherapy retroversion may lead to the presence of mature teratoma in metastatic foci [53].
Mature Teratoma
Mature teratomas are almost always cystic but 1 % may be solid. Mature cystic teratomas, also called dermoid cysts, are the most common germ cell tumours of the ovary [8], more common in young but also seen in older women. Histogenetically, they arise from a single germ cell after the first meiotic division and develop a diploid karyotype, which points to a parthenogenetic mechanism of histogenesis. They have a benign clinical course, in comparison to testicular mature teratomas which are aneuploid and malignant, although very rarely they may undergo malignant somatic mutation of one of their components. They present with the symptoms of most germ cell tumours or they may be asymptomatic, presenting as an incidental finding during other investigations. Rokitansky protuberances, the presence of fat and teeth, are diagnostic features on imaging.
Mature cystic teratoma is composed of a thin-walled cyst that usually contains hair and sebum. It may show focal thickening of the wall with calcification (Rokitansky protuberance), which should be sampled thoroughly as it may harbour different types of tissues. The cyst wall is usually lined by skin and its adnexa. Often, there is a foreign body giant cell reaction with cholesterol clefts. Other ectodermal derivatives present are mature neuroglial tissue and ocular structures including retina. Mesoderm may be represented by islands of mature cartilage, fat and smooth and skeletal muscle. Endoderm derivatives may be encountered in the form of respiratory or intestinal epithelium among others [4]. Immature tissue may be present, but if this is limited (less than 21 mm2) the prognosis is still excellent. Rarely, well-formed embryonal structures may be seen within a teratoma and this is termed teratoma with fetiform features (homunculus). It should be differentiated from fetus in fetu, which is retroperitoneal and derives from a monozygotic diamniotic twin.
Mature Cystic Teratoma with Malignant Transformation
Malignant transformation in a mature cystic teratoma occurs in up to 2 % of cases and this makes the prognosis unfavourable. It is more often encountered in older women. The most common tumour type is squamous cell carcinoma, but other malignancies have also been reported such as mucinous carcinoma, neuroendocrine tumours (carcinoid), thyroid carcinoma, basal cell carcinoma, malignant melanoma and sarcomas [2, 54–56]. Of interest, somatic transformation in testicular teratomas usually leads to a sarcomatous rather than carcinomatous malignancy [2]. These malignancies are characterised by wide local spread rather than metastatic nodal disease, contrary to the rest of the malignant germ cell tumours. The best prognostic scenario is squamous cell carcinoma within a mature cystic teratoma without any capsular breach. Treatment is total abdominal hysterectomy and bilateral salpingo-oophorectomy. Somatic transformations in teratomas are more often seen in older women, at an age where fertility issues are less commonly relevant. These tumours do not respond well to chemotherapy [54].
When tumours with mucinous features arise within a teratoma, these are likely to be of germ cell and not surface epithelial origin. They can have a spectrum of appearances ranging from cystadenomas, borderline tumours, to adenocarcinomas with intestinal phenotype. The presence of a teratomatous component excludes the possibility of metastatic spread from a gastrointestinal primary [57]. Only a small number of such cases have been reported and the prognosis has been good for cystadenomas and borderline tumours and variable for adenocarcinomas. Occasional tumours present with features of pseudomyxoma peritonei [58].
Monophasic Teratoma
Struma Ovarii
Monophasic teratomas are those which are entirely or almost entirely composed of derivatives of one germ cell layer. The most common is thyroid tissue in the ovary, known as struma ovarii (Fig. 5.5). It may be entirely composed of benign thyroid tissue and it may present as a palpable abdominal mass or rarely with features of Meigs syndrome simulating ovarian carcinoma [59]. Struma ovarii may show features of a follicular adenoma or carcinoma [60], and the distinction is difficult because usually the lesion is not encapsulated and the diagnostic criteria of capsular invasion cannot be applied. The diagnosis of follicular carcinoma is made when there is lymphovascular invasion or infiltration of normal ovarian parenchyma present. Papillary thyroid carcinoma may also arise in the background of struma ovarii and constitutes the most common malignant change. Occasionally, benign thyroid tissue peritoneal implants may be present (struma peritonei) and this does not have any clinical significance with regard to prognosis, although this is controversial owing to the rarity of such cases [61].
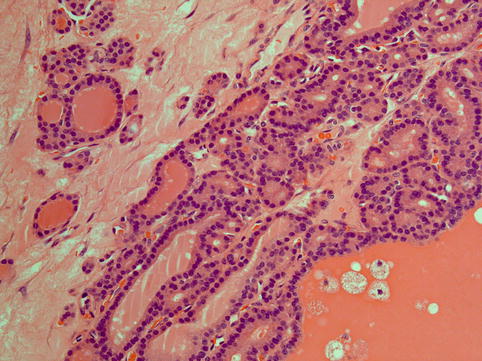
Fig. 5.5
Struma ovarii is composed of a sole, predominant or nodule-forming component of mature thyroid tissue characterised by colloid-filled follicles lined by cuboidal cells
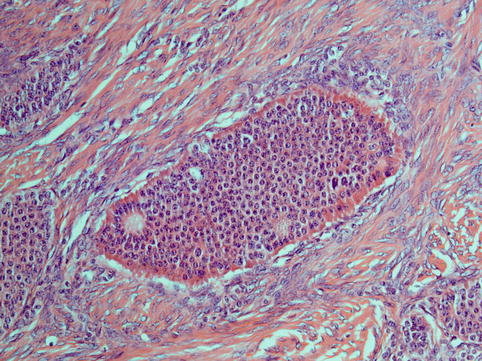
Carcinoid
In other parts of the body, the term carcinoid is tending towards becoming obsolete in favour of the broader term neuroendocrine tumour and further qualifiers describing its biological behaviour, as exemplified by terminology used in lung and gastrointestinal pathology. However, a broad consensus has not been established and the term is retained in the WHO classification of tumours of the female genital tract [4]. Ovarian carcinoids may be pure or part of a dermoid cyst. They are rare and may present with carcinoid syndrome. A diagnostic feature is elevated serum 5-hydroxy-indolacetic-acid (5-HIAA) levels. These are tan to yellow firm nodules grossly and they resemble gastrointestinal carcinoids microscopically. The tumour cells show neuroendocrine features which consist in round central nuclei with a granular (‘salt and pepper’) chromatin pattern and eosinophilic granular cytoplasm due to the presence of neurosecretory granules (Fig. 5.6). They are distinguished into insular or trabecular types [62–64], depending on how the tumour cells are arranged, strumal carcinoids if they are associated with struma ovarii and mucinous (goblet cell carcinoid, a distinct type of tumour classified traditionally as a carcinoid, which is much more aggressive than a common carcinoid). Insular carcinoid is the most common primary ovarian carcinoid and is considered to be malignant [62]. It is usually seen in older women, and total abdominal hysterectomy and bilateral salpingo-oophorectomy is the treatment of choice. There is little experience with chemotherapy in the treatment of these tumours. Trabecular carcinoids of the ovary should be distinguished from metastases. They are called trabecular because of a ribbon-like architectural pattern. They are not associated with metastases and they are treated surgically with follow-up and no further treatment [63]. Strumal carcinoid usually behaves in a benign fashion [65]. Goblet cell carcinoid is much more aggressive, similar to its appendiceal counterpart, it is associated with lymph node spread and it is preferentially treated with hysterectomy and bilateral salpingo-oophorectomy when maintenance of fertility is not a relevant issue [66].
 Psychological Aspects of Hereditary and Non-hereditary Ovarian Cancer
Psychological Aspects of Hereditary and Non-hereditary Ovarian Cancer
 Management of Patients with Early-Stage Ovarian Cancer
Management of Patients with Early-Stage Ovarian Cancer
 Ovarian Cancer Screening and Early Detection
Ovarian Cancer Screening and Early Detection
 Diagnosis and Management of Epithelial Ovarian Cancer with Peritoneal Metastases
Diagnosis and Management of Epithelial Ovarian Cancer with Peritoneal Metastases
 Treatment of Advanced Stage Ovarian Cancer
Treatment of Advanced Stage Ovarian Cancer
 Surface Epithelial Tumours of the Ovary
Surface Epithelial Tumours of the Ovary




Related posts:
Psychological Aspects of Hereditary and Non-hereditary Ovarian Cancer
Management of Patients with Early-Stage Ovarian Cancer
Ovarian Cancer Screening and Early Detection
Diagnosis and Management of Epithelial Ovarian Cancer with Peritoneal Metastases
Treatment of Advanced Stage Ovarian Cancer
Surface Epithelial Tumours of the Ovary
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
