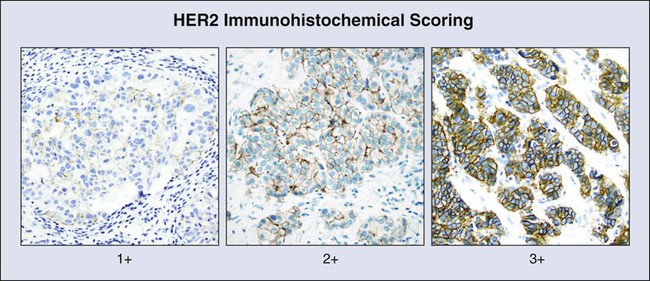
Wilbur A. Franklin, Dara L. Aisner, Miriam D. Post, Paul A. Bunn and Marileila Varella Garcia • Biomarker development has profoundly affected basic understanding of carcinogenesis and expanded means of intervention in human cancers. Biomarkers have been applied with variable success in three broad areas that correspond to phases of tumor development and progression: (1) early detection, (2) diagnosis, and (3) prediction of clinical outcome (prognosis) and response to targeted treatment. • For early detection, biomarkers go through five stages of development: (1) identification of promising directions; (2) validation of a clinical assay; (3) demonstration that the biomarker can detect disease before it is clinically relevant; (4) evaluation of the biomarker during prospective screening; and (5) quantification of the effect of screening on reducing the burden of disease in a population. • Early detection using quite different approaches has changed the natural history of cervical, colorectal, and lung cancer. Progress depends on improving our understanding of the biology of site-specific carcinogenesis and using intervention strategies informed by basic biology. • Cancer diagnosis still relies on an evaluation of tumor tissue histology, a complex process associated with interindividual variation; however, new approaches to automated quantitative histologic evaluation and immunohistochemical analyses are under active development. • New molecular diagnostic approaches that utilize assessment of quantitative messenger RNA signatures, fluorescent-in-situ hybridization, quantitation of microRNAs, and genetic sequencing can now be accomplished with accurate use of formalin-fixed paraffin-embedded tissues. • Molecular testing can provide guidance in prioritizing therapies for patients most likely to benefit and can identify patients who have been proven not to benefit from a targeted therapy. • Molecular testing can also provide clinicians with a rationale to guide specific patients toward specific clinical trials. • As new molecular techniques have been applied to cancer diagnosis and treatment, the necessity of developing standardized processing approaches for solid tumor specimens has been appreciated and has helped to change tissue collection and specimen-handling routines. Since the time of Virchow more than 150 years ago, diagnosis and treatment of cancer have depended on the appearance of tumor cells under the microscope and the diverse and often subtle morphologic differences that distinguish benign from malignant cells. Careful study of cellular features and later of molecular features of both hematologic and solid tumors has led to the idea that cancer results from a series of genetic and phenotypic changes in progenitor cells that occur over a period of many years. In epithelial tumors, the final histologic changes that define transformation of premalignant lesions into malignancy are the invasion of stromal tissues, the elicitation of a stromal response, including angiogenesis, and metastasis to lymph nodes and distant sites.1 The clinical implications of this multistep process are best illustrated in the cervix and colon, where early intervention has resulted in reduced mortality from cancer. Once invasive carcinoma has occurred, testing becomes more urgent and intervention more aggressive. This chapter reviews concepts of solid tumor carcinogenesis and biomarker development for early detection and distinguishes early detection testing from biomarker applications in diagnosis, prognosis, and treatment of established solid tumors. Hematologic biomarkers are described elsewhere in this volume. Finally, current best practices in molecular testing are discussed. Remarkable strides have recently been made in reducing morbidity and mortality through early intervention in several different kinds of solid tumors. The triggers for intervention have until now been largely based on cytologic evaluation of exfoliated cells or advances in imaging technology. To date, few molecular markers have been sufficiently sensitive, specific, or accessible to be useful in population-based studies for early detection or prevention studies. Reasons for this lack of usefulness are numerous. Sensitive biomarkers for risk assessment and early detection must be inexpensive, noninvasive, and easily applied to be useful for population-based screening. However, such biomarkers are rarely sufficiently specific for diagnosis, prognosis, or response to treatment. A formal process to guide the development of early detection biomarkers has been defined by Pepe et al.2 In this construct, biomarkers must pass through five phases of development that correspond to the strength of evidence that the biomarker will be a useful population screening tool. These phases of development are analogous to the structure for therapeutic drug development3 that has been used for many years. The phases defined by Pepe and colleagues are listed in Table 17-1 and are the framework for the discussion in this chapter of biomarker development in individual organs. Table 17-1 Phases of Biomarker Development by Pepe and Colleagues Invasive cervical carcinoma is one of the few tumors that results from well-defined premalignant lesions that can be identified and effectively treated, reducing morbidity and mortality from a tumor that is the third most common in women worldwide.4 More than 60 years ago, it was recognized that cellular abnormalities in cervical epithelium detectable in exfoliated cells could predict the presence of carcinoma.5 By 1988, premalignant cervical cytology had been codified as the Bethesda System for reporting cervical/vaginal cytologic diagnoses.6 Population-based light microscopic screening for atypical cells in cervical smears with surgical removal of detected abnormal epithelium in Great Britain is estimated to have reduced the prevalence of invasive squamous carcinoma by 80% after 1988.7 Conventional cervical cytology is thus one of the most effective early detection biomarkers in use today. Delineation of molecular carcinogenesis in the cervix (Figs. 17-1 and 17-2) has provided alternative methods for detection, risk assessment, and diagnosis. Nearly all cervical carcinomas and their precursor lesions are caused by human papillomavirus (HPV).8 HPV is a DNA tumor virus that in 1934 was shown by Peyton Rous to produce squamous tumors in rabbits.11–11 It was not until 1974 that zur Hausen recognized the association between HPV and cervical cancer.14–14 Persistent infection with high-risk strains of HPV, most commonly type 16 and 18,15 is an essential step in cervical carcinogenesis (reviewed in reference 16).16 The virus encodes three proteins that stimulate cell proliferation and survival: E5, E6, and E7 (reviewed in reference 17).17 The viral proteins E6 and E7 interfere with p53 and Rb, respectively,18,19 resulting in resistance to apoptosis and chromosomal instability. The HPV genome may be integrated into the host cell genome, accounting for long-term persistence of the virus. Integration of E6 and E7 genes20,21 is crucial to progression of the infected cell to a malignant state, whereas other viral genes may be lost. Detection of HPV DNA in clinical samples can be accomplished by a number of methods, with the dominant techniques based on polymerase chain reaction (PCR) or hybridization with signal amplification (reviewed in reference 22).22 In the United States, the predominant assay is the Food and Drug Administration (FDA)-approved Hybrid Capture 2 method (Qiagen, Germantown, Md.). In this technique, viral DNA hybridizes with an HPV RNA probe cocktail that detects multiple high-risk HPV types. The RNA : DNA hybrid is captured by hybrid-specific antibody that coats the surface of a microplate well. The hybrids are detected by a second hybrid-specific antibody conjugated to alkaline phosphatase. Each hybrid may bind to multiple antibodies, amplifying signal. Antibody-conjugated alkaline phosphatase cleaves a chemiluminescent substrate, emitting a light signal that is quantified with use of a luminometer. Light emitted above a standardized threshold intensity indicates the presence of virus. The test covers 11 HPV types in addition to HPV 16 and 18 and has a reported analytic sensitivity of 1.09 picograms/mL. A reengineered version of this hybrid capture test for low resource settings, careHPV, covers 14 HPV types, requires no running water, and has a turnaround time of 2.5 hours. The test has been successfully used in rural China with a clinical sensitivity of 90% for the detection of cervical intraepithelial neoplasia types 2 and 3.23 Understanding of the molecular oncogenesis of cervical squamous carcinoma has provided a standardized biomarker of risk. More than 95% of HPV infections clear up without further consequences. However, persistent infection for months to years with HPV type 16 or 18 carries a 40% risk of squamous intraepithelial lesions (SILs),24 and untreated high-grade SILs have been estimated to result in invasive carcinoma in approximately 30% of persons over 5 to 10 years.24,25 Persistent HPV infection thus indicates high risk for eventual invasive carcinoma and represents an additional biomarker that could be useful in the prevention of invasive carcinoma. Testing for HPV DNA has been found to be more sensitive than cytologic screening for detection of high-grade SILs26,27 but somewhat less specific. In view of the number of infections that are self-limited, relying on a single HPV test may result in greater numbers of negative colposcopy examinations. To address this problem, co-testing strategies have been evaluated whereby both cytology and HPV testing are performed on initial examination.28 Negative results have a high negative predictive value, and the rescreening interval can be extended from 1 to 3 years, reducing overall screening expense. Persons with positive results of a viral DNA test but negative cytologic findings have been rescreened at 1 year and examined with colposcopy if positive results are found. Whether this strategy or other strategies for cervical cancer risk management will be universally adopted remains to be determined.22,29 What is clear is that HPV testing provides a means for improving sensitivity for detection of early cervical lesions, further reducing mortality from invasive cervical carcinoma while potentially reducing screening expense. Finally, HPV is not only a biomarker for risk assessment and colposcopic intervention but is an immunogen that has been successfully incorporated into bivalent (types 16 and 18, Cervarix) and quadrivalent (types 6, 11, 16, and 18, Gardasil) vaccines to prevent infection and carcinogenesis (reviewed in references 30 and 31).30,31 An in-depth discussion of HPV vaccines for tumor prevention is beyond the scope of this chapter. However, the many facets of HPV expression in cervical carcinoma and the emerging role of HPV in other tumors, including vulvar and vaginal condylomata, certain skin cancers, and head and neck carcinoma, illustrate how a valuable biomarker that is integral to oncogenesis can be used to improve the accuracy of estimating tumor risk and the specificity of treatment. Unfortunately, such clearly defined and easily accessible biomarkers are rare. Rather, the more usual case is described in the next sections for colon and lung, where potential biomarkers are defined but have not found a role in early detection, diagnosis, prevention, or treatment. Like cervical carcinoma, colonic adenocarcinoma can be effectively prevented by extirpation of premalignant epithelium, which in the colon is represented by polyps. Several recent trials have indicated that removal of polyps by colonoscopy34–34 reduces the frequency of colorectal carcinoma (CRC), and consensus opinion is that endoscopic polypectomy has a significant impact on the incidence of CRC. Recent data have also suggested that colonoscopic screening reduces CRC mortality by more than 50%34 and that the less extensive sigmoidoscopy reduces mortality from distal CRC by a nearly equal percentage (50%).35 The latter sigmoidoscopic study was a large, randomized, population-based screening trial. Colonoscopic screening for colon polyps beginning at age 50 years has become a standard of care. Molecular biomarkers to date have not played a direct role in reduction of morbidity and mortality in persons with CRC. However, molecular studies have facilitated understanding of colon carcinogenesis (Figs. 17-3 and 17-4) and provided a rationale for colonoscopic surveillance and polypectomy. At the mid twentieth century, an association between benign polyps was suspected, but at that time no direct evidence was available to show that polyps undergo transition to CRC.36 Suspicions were finally confirmed in the 1980s, when the clonal nature of both sporadic polyps and CRC was documented.37 Activating mutations in the KRAS oncogene and inactivating mutations in several tumor suppressor genes were described.38 The same mutations were demonstrated in both polyps and carcinoma, and the number of mutations was found to increase with the histologic grade of the adenoma. This finding suggested a progression to invasive carcinoma through the accumulation of genetic and epigenetic changes. These changes provide a proliferative and survival advantage to mutant cells that continue to proliferate and mutate until a malignant phenotype is reached. The combined data from these studies led to the proposal of a multistep model of colon carcinogenesis39 that continues as the standard model.40 Several forms of hereditary colon cancer are recognized (reviewed in reference 41)41 and are listed in Table 17-2. Table 17-2 The first hereditary colon carcinoma to be described, familial adenomatous polyposis (FAP), is characterized in its classic form by the occurrence of hundreds to thousands of polyps in the colon of affected persons with the formation of invasive carcinomas at an early age. Polyps may also sometimes be found in the upper gastrointestinal tract. As long ago as 1882, siblings were described with numerous and recurring polyps,42 and by the mid twentieth century, FAP was established as a hereditary autosomal-dominant disorder.43 A gene mutation associated with FAP was identified on chromosome 5q by linkage analysis44,45 and positional cloning studies.46–49 The gene, adenomatous polyposis coli (APC), is now known to encode a tumor suppressor that modulates wnt signaling through binding and stabilization of a complex of proteins that includes GSK3b, β−catenin, and axin family members. Mutations in APC truncate the protein at its carboxyl terminus and disrupt the protein signaling complex, which results in deregulation of cell cycle control, cell migration, differentiation, and apoptosis (reviewed in reference 50).50 In addition to being responsible for FAP, APC mutation is present in approximately 60% of sporadic colon adenocarcinomas51,52 and an approximately equal number of sporadic polyps,51,53 suggesting that APC mutation is an early event in sporadic carcinogenesis, as well as in hereditary disease. An attenuated form of FAP with smaller numbers of tumors (<100) has been described.54,55 This condition is referred to as attenuated FAP54 and may be driven by the mutations in APC in the 3′ and 5′ ends rather the central portion of the gene that is affected in classical FAP.55 The most common form of hereditary colon cancer is hereditary nonpolyposis colon cancer (HNPCC). HNPCC is a familial syndrome that differs from FAP in that it manifests a smaller numbers of polyps compared with FAP. The earliest description of this disorder can again be traced back to the early twentieth century,56 but the hereditary character and syndromic features of the disease were firmly established by the diligent kindred studies of Lynch and Lynch,57 after whom the syndrome is named (Lynch syndrome). In brief, the syndrome consists of CRC that occurs in close relatives, often younger than 50 years,58,59 without adenomatous polyposis. HNPCC is also associated with an increased frequency of tumors in the endometrium, stomach, ovary, hepatobiliary tract, upper urinary tract, small bowel, and central nervous system.41 In contrast to FAP polyps, which are frequently found on the left side of the colon, persons with HNPCC frequently have flat lesions on the right side of the colon with a distinct histologic appearance referred to as “serrated adenoma” (Fig. 17-3). The molecular genetic underpinnings of HNPCC were deciphered beginning with the linkage to microsatellite markers on chromosome 2p.60 The genetic basis of the disease was established when it was found that patients with the syndrome harbored mutations in the mismatch repair (MMR) genes hMLH1,61,62 hMSH2,63,64 hMSH6,65 and hPMS2,66,67 with mutations in hMLH1 and hMSH2 accounting for more than 90% of HNPCC cases. The germline mutations are heterozygous, and colon carcinogenesis results from silencing of the nonmutant allele by methylation68,69 or by acquired allelic loss. The phenotypic consequence of gene silencing is loss of MMR protein and the resulting inability to repair DNA replication errors, ultimately leading to accumulation of mutations contributing to carcinogenesis. At a clinical level, protein loss can be the demonstrated by immunochemistry and optimally requires four separate stains. The inability of affected cells to repair DNA replication errors results in insertions or deletions in short tandem repeats (microsatellites) that exist throughout the genome and is referred to as “microsatellite instability” (MSI).70,71 Currently, MSI is documented by testing of a standard set of five microsatellite markers recommended by a consensus panel.72 Results are scored on the basis of the number of unstable loci and are classified as MSI-High (MSI-H), MSI-Low (MSI-L), or MSI-Stable (MSI-S). More than 90% of HNPCC tumors are MSI-H, but approximately 15% of all sporadic tumors are also MSI-H,73 and thus the test is not specific for hereditary disease. A third hereditary variant of colon carcinoma is MUTYH-associated polyposis (MAP), which is the newest polyposis syndrome (first described in 2002).74 MAP is a disorder similar to attenuated FAP and is characterized by adenomatous polyps that may number in the hundreds but not in the thousands, usually ranging from 10 to 1000.77–77 Polyps may also occur in extra colonic sites, usually the duodenum. Adenomas tend to occur in the proximal colon.76 MUTYH is a glycosylase and part of the base excision repair pathway. Oxidative damage to DNA frequently results in the modification of guanine (G) to 8-oxoG. 8-oxoG can pair with adenine (A) during DNA replication, a mismatch that is recognized by MUTYH, which removes the mismatched A and restores the correct base, cytosine (C). The damaged G (8-oxoG) is then replaced with an intact G. Mutation of MUTYH at specific hot spots hinders recognition of the 8-oxoG : A mispairing, resulting in G : C to A : T transversion at sites of oxidative damage. The mutational hot spots are found in exon 7, resulting in amino acid substitution Y179C, and in exon 13, resulting in substitution G496D. Germline mutation at both hot spots (biallelic mutation) occurs in 0.3% of patients with CRC. Biallelic mutation confers 28-fold relative risk for CRC compared with control subjects. Monoallelic mutation is more common with a frequency of approximately 2% in CRC but also occurs in approximately the same frequency in control populations. Most studies suggest that monoallelic mutation confers little or no increased risk of CRC.75,76 Among control patients without polyposis, biallelic mutation is rare.76 The fourth hereditary form of colon cancer is the hamartomatous polyposes, a category that includes Peutz-Jeghers syndrome (PJS) and juvenile polyposis syndrome. First described in the 1890s,78,79 PJS was named in 1954 by Bruwer and colleagues83 in recognition of the work of Peutz80 and Jeghers.81,82 PJS is an autosomal-dominant condition characterized by multiple histologically distinct polyps (Fig. 17-3) that occur in the large and small bowel in association with melanin pigmentation of the lips, mouth, nose, anus, and extremities.84 The condition is associated with a high risk for cancer in not only the large and small intestine but also in the esophagus, stomach, pancreas, breast, uterus, ovary, and lung.87–87 Gene mapping has identified a locus on chromosome 19.3 p1388,89 that encodes a serine-threonine kinase, STK11 (LKB1), a gene that is mutated in more than 90% of affected individuals.90–94 A variety of mutations including deletions, truncations, and missense point mutations may occur over nearly the full coding region of the gene.86 STK11 protein is multifunctional, playing a role in cell cycle arrest, wnt signaling, and the tuberous sclerosis complex pathway, with effects on mammalian target of rapamycin signaling.84 Mutation, often associated with loss of heterozygosity,95,96 results in loss of function, and the gene is thus considered to be a tumor suppressor gene. The overall frequency of PJS is estimated at 0.5 to 2.0 per 100,000 live births.97 Juvenile polyps are characteristically solitary peculated lesions that contain tortuous, often cystically dilated crypts, stromal granulation tissue and inflammation, and denudation of the surface epithelium.98 These lesions frequently occur in children, and there is no evidence that solitary juvenile polyps pose an increased of risk for carcinoma.100–100 However, multiple juvenile polyps may occur in young persons and their family members,103–103 and the presence of >5 juvenile polyps or a single juvenile polyp in a person with a family member with juvenile polyposis now defines the condition, termed “juvenile polyposis.” This form of polyposis carries a lifetime risk of colon carcinoma of 39% and a relative risk of 34.104 Fifty percent to 60% of affected individuals carry germline mutations in SMAD4 or BMPR1A.105–107 Nearly all patients with SMAD4 mutations also have hereditary hemorrhagic telangiectasia108 (overlap syndrome). Both SMAD4 and BMPR1A are involved in the transforming growth factor–β (TGF-β)/bone morphogenetic protein pathway. Finally, a variety of polyposis syndromes with potential genetic linkage have been described and are best exemplified by hyperplastic polyposis syndrome (serrated polyposis syndrome).109,110 This condition consists of the occurrence of multiple sessile polyps variously referred to as hyperplastic polyps or serrated adenomas, frequently on the right side of the colon and numbering from 5 to >100.111 Although familial associations of the syndrome112 and linkage to high risk of colon carcinoma113 have been reported, the genetic basis is complex and associated with mutations in several driver genes, including BRAF and KRAS.110 No single genetic mechanism has been linked to the syndrome, and the true frequency is not established. To date, the number of reported cases is small. Although the number of highly penetrant mutations that predispose to colon carcinoma is large, the proportion of colon carcinomas that can be accounted for by these mutations is approximately 5%41 and has been overestimated in the past.73 Markers of hereditary predisposition and molecular progression in sporadic tumors that have been delineated to date have not been easily converted into an early detection test suitable for broad population-based applications for several reasons. First, the types of accessible specimens that can be used for tests for early detection consist primarily of blood and fecal DNA. Although DNA from invasive tumors can be detected in the blood by PCR-based mutation testing, it is not as useful at earlier polyp stages. In addition, collection and processing of fecal specimens for a biomarker that is expected to be present at a low level and heavily contaminated by nonneoplastic cells is a logistical challenge. Second, some of the genetic abnormalities identified in colon carcinogenesis do not readily lend themselves to screening assays. Mutations in APC, for example, consist mainly of truncations and deletions that may occur over a wide region of the gene, and thus designing a sensitive clinical assay has been difficult. Finally, the wide availability of colonoscopy services that combine polyp detection and excision reduces the cost and improves the attractiveness of direct and systematic colonoscopic population-based surveillance. Biomarkers that have been most successful for colon cancer screening are fecal tests such as the guaiac fecal occult blood test (FOBT), the fecal immunochemical test, and the stool DNA (sDNA) test. The greatest experience has been with the FOBT. This test is based on a color change whereby α-guaiaconic acid extracted from the wood of tropical Guaiacum tree is rapidly oxidized by hydrogen peroxide to a blue-colored quinone in the presence of heme derived from blood. A wide variability in results occurs114 as a consequence of the level of compliance with collection methods, the type or brand of kit used, the frequency of testing, user interpretation, and other factors. Despite a lack of standardization, FOBT screening with follow-up colonoscopy is reported to reduce both incidence115 and mortality116,117 from colon carcinoma. The former is attributed to polyp removal. The fecal immunochemical test is a second test for occult blood and is reported to be more sensitive and specific than the guaiac assays. It has been shown to have a cancer detection rate similar to colonoscopy but has a lower sensitivity for polyps.118 Occult blood tests measure host response (hypervascularity and bleeding) to a tumor. The sDNA test, however, aims to measure a direct product of tumor cells. A recent version of the sDNA test measures multiple markers in stool using an array-based platform119 and for this reason has been referred to as “next generation,” although this technology does not use deep sequencing methods and should not be confused with “next generation sequencing.” The biomarkers that are incorporated into the platform include four methylation probes, as well as mutated KRAS and hemoglobin. Patterns of gene methylation have been identified in colon tumors that have been characterized as CpG island methylation high and CpG island methylation low (reviewed in reference 120).120 When applied to a preliminary cohort of 678 subjects with polyps or colorectal carcinoma, the combination test including methylation markers, KRAS mutation, and occult blood had a sensitivity of 85% for carcinoma and 54% for polyps. Despite the availability of these fecal-based tests, the target in early colon cancer intervention appears to be shifting from detection of invasive carcinoma to prevention by detection and thorough clearing of adenomatous polyps from the colon. Defining the biology of colon carcinogenesis and confirming that polyps are the precursor lesions along with improvements in colonoscopic methods has led to reduction in mortality from colon cancer. Colonoscopy enables direct identification of potential precursor lesions and at the same time provides a means for ablating them, which is strikingly evident in the new sigmoidoscopy findings,35 indicating that although sigmoidoscopy reduces mortality from visible distal colon tumors, mortality from proximal lesions is unaffected. It seems likely that colonoscopy will be the main vehicle for early detection and intervention in colon cancer for the foreseeable future. For several decades, lung carcinoma has had the highest mortality of all tumors, accounting for more than twice as many deaths as the next most lethal tumor.121,122 Initial large-scale efforts during the 1970s and 1980s to reduce mortality by early detection using chest radiographs and/or sputum cytology were not successful. Chemoprevention using β-carotene and/or alpha tocopherol dietary supplementation also failed.123–126 Recently, however, early detection efforts have begun to yield some success in reducing mortality even in this problematic tumor. Low-dose computed tomography (LDCT) has a higher clinical sensitivity for detecting smaller tumors than does a chest radiograph. Early studies in smokers documented a 2.7% prevalence of malignant disease detected by LDCT versus 0.7% in the same patients screened by chest radiograph.127 In a multicenter study of more than 30,000 smokers, LDCT detected lung cancer in 1.3% at baseline and an additional 0.3% at annual follow-up examinations for an overall total of detection rate of approximately 1.5%.128 Although this study was not randomized and had no unscreened control arm, it showed a survival advantage in comparison with historical control subjects. Recently, a large randomized trial of CT versus radiography conducted annually in three rounds found that LDCT detected carcinoma in 2.4% of the screened high-risk population and resulted in a 20% reduction in lung cancer mortality that was not observed in the chest radiograph control group.129 The weight of evidence supporting improved long-term mortality rates in smokers has prompted the publication of multisociety consensus guidelines recommending annual LDCT screening for smokers and former smokers ages 55 to 74 years with access to appropriate follow-up.130 Unlike mortality reductions that have been achieved in the colon and cervix, where preinvasive and therefore nonmalignant lesions are targeted, the benefits of LDCT screening for lung cancer have been achieved through early detection of invasive tumors. A strategy to identify in situ premalignant lesions could potentially reduce lung cancer mortality to a much lower rate. Unfortunately, what are thought to be premalignant lesions in the lung are difficult to detect and may occur at unpredictable locations anywhere in the central and alveolar airways (Figs. 17-5 and 17-6), exemplifying the challenges confronting prevention efforts in some organ systems. The predominant premalignant lesion of the central airways is squamous dysplasia131 (also referred to as “atypia”). Squamous dysplasia was documented more than 50 years ago in association with carcinoma,132 as well as in smokers without concurrent carcinoma.133 Central squamous lesions are flat, inconspicuous, and only rarely identified by white light fiberoptic bronchoscopy. They are somewhat better visualized by laser-induced fluorescence emission bronchoscopy.134 This method relies on a loss of autofluorescence, and signal from dysplasias may be mimicked by fluorescence loss caused by mucosal hemorrhage and other reactive changes. Thus, although sensitivity of laser-induced fluorescence emission bronchoscopy is high, specificity is low. Prospective studies on the power of dysplasia to prospectively predict future central airway malignancy are few and face the forbidding challenges of inaccessibility of the bronchi and the prolonged follow-up that is required to achieve significant results in a sufficiently powered cohort. Sputum studies indicate that atypia of cells exfoliated from the central airways is associated with elevated risk for incidental carcinoma. The hazard ratio increases with the proximity of the sputum collection date to lung cancer diagnosis, but sputum atypia even 3 years prior to lung cancer diagnosis is associated with increased risk of lung carcinoma.135 Confirmation of sputum analyses in biopsy studies to identify sources of dysplastic epithelium will require a large and disciplined multicenter trial involving bronchial mapping and revisiting dysplastic sites to evaluate the long-term disposition of discovered lesions. The corollary question of whether ablation of premalignant central airway lesions could reduce lung cancer mortality remains an open one. A recent chemoprevention study136 using Iloprost, a prostacyclin agonist, has suggested that reversal of premalignant dysplasia morphology may be possible, but the study was a small one, and the end point was purely histologic. The long-term effect on mortality of agents that may alter the premalignant morphology of central airway epithelium remains unknown. As in other organs, most notably the colon, molecular changes that accompany the morphologic changes of dysplasia have been investigated in considerable detail. These studies are informed by the well-established role of tobacco smoke as a carcinogen in lung cancer. The dominant carcinogenic compounds found in cigarette smoke are the polyaromatic hydrocarbons and nicotine-derived nitrosamine ketone.137 These compounds are metabolized to highly reactive intermediates that are inactivated largely by mitochondrial glutathione transferases. However, repair is imperfect, and a small proportion of the intermediates react directly with DNA, forming bulky adducts. Unless removed by the DNA repair system, adducts can create errors of replication. Over time, molecular alterations accumulate, providing a selective advantage to mutant cells for growth and survival. Components of this pathway have been associated with increased risk for future lung cancer. Case control studies indicate that current smokers with tumors have higher levels of adducts in blood or tissue but that nonsmokers and former smokers do not have these higher levels.138 Bulky adducts appear to reflect ongoing smoking-related DNA damage rather than biological changes specifically associated with malignant transformation. Clinically applicable adduct levels predictive of lung cancer have not been established. Glutathione transferase and DNA repair enzyme mutations and polymorphisms that affect DNA repair activity could represent a risk factor for lung cancer because efficient deactivation of reactive carcinogens and repair of DNA damage caused by bulky adducts are important in protecting potential precursor cells from critical mutational changes. However, single-nucleotide polymorphism (SNP) association studies in many carcinogen metabolizing and DNA repair enzymes have demonstrated only weak and inconsistent associations and have not been sufficiently informative to guide intervention in potentially affected persons.139–143 DNA damage in lung premalignancy engenders a number of genetic and epigenetic changes that provide a cellular survival or proliferative advantage to affected bronchial epithelial cells. Perhaps the earliest molecular events may be epigenetic and include DNA methylation, histone modification, and alterations in micro (mi)RNA expression (reviewed in references 144 and 145).144,145 The greatest experience regarding the use of epigenetic changes in the context of early detection of lung cancer has been obtained in regard to methylation. Methylation refers to the conversion of cytosine to 5′ methylcytosine by DNA methyltransferase. In lung carcinogenesis, methylation preferentially occurs in CpG-rich regions (CpG islands) that are present in normally unmethylated promoter regions of critical genes (hypermethylation) and may silence gene expression. In lung tumors, interest has focused on genes such as p16,146 which are silenced in tumors but expressed in normal control tissue and are considered to be tumor suppressor genes (TSGs). The development of a simple and sensitive assay based on the conversion of methyl cytosine to uracil in the presence of metabisulfite followed by PCR with primers specific for the methylated bases, termed methylation-specific PCR,147 has greatly facilitated the assessment of methylation status as a risk marker for lung carcinoma. Methylation of key TSG promoter regions in cells exfoliated from bronchial surfaces into the sputum is nearly universally associated with prevalent lung carcinomas148 and frequently occurs in sputa of smokers without a tumor as well. TSG methylation is not present in sputum of people who have never smoked. This finding initially led to optimism that methylation could be used to screen selected populations at risk for lung carcinoma. However, the predictive power of single gene methylation or methylation of a small number of genes is limited by the ubiquity of methylation in smokers and the consequent high rate of false-positive results. This problem has led to the strategy of testing promoter regions in large numbers of genes to estimate overall methylation burden.149 Identification of panels of genes to optimize the predictive power of methylation testing is a process that is still under way. In receiver-operator curve analyses, a seven-gene methylation profile selected from a 31-gene panel achieved a predictive accuracy of 71% to 77%. However, with a sensitivity set at 75%, the false-positive rate was 75%.150 Although TSG promoter methylation may prove problematic as a general screening tool, it may be useful in specific niche populations or to supplement other methods of screening such as LDCT, and ongoing studies aim to further evaluate the usefulness of this approach. A second molecular consequence of tobacco exposure is chromosomal instability, which is detected as aneuploidy by fluorescence in situ hybridization (FISH) in bronchial epithelial cells in situ and in sputum. Aneuploidy is nearly universal in lung carcinoma. It is also frequently observed in high-grade dysplastic lesions in the central airways.151 Aneuploidy in sputum has been associated with prevalent carcinoma152,153 and has recently been shown to have a sensitivity of 76% within a detection window of 18 months before the appearance of invasive carcinoma.154 However, FISH performed on sputum is labor intensive and difficult to perform both in the specimen preparation and slide interpretation. FISH requires a high level of expertise at every step, which has led to the development of automated methods of FISH screening. However, to date, these automated methods have not been broadly applied. It seems likely that FISH may most successfully be applied in restricted settings in conjunction with other technologies. High-throughput methodologies have been suggested as possible early detection tools. Recently, genome-wide association studies have identified an SNP variant at 15q15.2 that is significantly associated with cancer risk, with an overall odds ratio of 1.15.155 The low odds ratio suggests that the SNP variant itself will not represent a useful predictor. However, this region of the gene still needs to be explored for more powerful biomarkers. Gene expression microarrays have identified consistent changes in the transcriptome of epithelium brushed from nonmalignant bronchial surfaces that distinguish smokers from nonsmokers.156 An additional set of changes are reported to identify subjects with cancer from smokers without cancer at an accuracy of 83%.157 Finally, large-scale genomic sequencing studies have identified differences in the “genomic landscape” between smokers and nonsmokers.158 It is expected that manageable numbers of biomarkers and clinically applicable testing platforms will emerge from these exploratory studies that will improve risk assessment and management of high-risk smokers, as well as patients with already invasive carcinomas. Premalignant cellular and molecular changes are described in several other tumors, including breast, prostate, bladder, and esophagus. A detailed review of these changes may be found in appropriate chapters of this text (see Chapters 74, 83, 84, and 91). However, identification of these changes has not led to mortality reductions. IHC staining for estrogen receptors (ERs) and progesterone receptors (PRs) was one of the earliest applications to find a place in patient management. Until the early 1980s, antibodies to these proteins were not widely available. However, with the development of monoclonal antibody technology, antibodies that could identify hormone receptors were created159 and found to reliably label receptors even in FFPE sections of breast carcinomas.160,161 Unexpected results of this testing were that these receptors are typically found in the cell nucleus and that the intratumoral and intertumoral range of expression is large. Because of this finding, semiquantitative scoring that takes into account intensity of staining and percentage of cells stained has been used to establish expression levels that define positive and negative results.162,163 These scoring systems continue to prove their prognostic value to the present time.164 A second IHC test that has been successfully applied to the management of breast carcinoma is the test for HER2/neu (ERBB2) expression. HER2 is a tyrosine kinase receptor (TKR) and a member of the epidermal growth factor receptor (EGFR) family of signaling molecules. Its role in human carcinogenesis was initially discovered through DNA screening, where it was found to be amplified in 30% of the tested breast cancers.165 RNA and protein were shown to be overexpressed in tumors that are amplified, as well as in a subset of nonamplified tumors.166 HER2 testing became crucial when it was found that HER2 protein overexpression was a determining factor for in vitro and xenograft responsiveness to anti-HER2 monoclonal antibody treatment.167,168 In early clinical trials, anti-HER2 IHC was used as the method for determination of HER2 status and determined eligibility for treatment with the anti-HER2 antibody drug, Herceptin.169 A standardized approach to HER2 IHC testing was established several years later when expert panels agreed upon criteria for determining HER2 status (Fig. 17-7).170 Recently, tests for multiple IHC markers have been combined into panels that include ER, PR, HER2, and the proliferation marker Ki-67,171 as well as several other markers thought to be of prognostic importance.172 These panels have been found to provide prognostic information that is comparable with that of multiplex mRNA expression assays discussed later and have the added advantage of accessibility in local laboratories and ready correlation with histologic features. A substantial subset (~15%) of breast tumors do not express ER, PR, or HER2 and are now referred to as triple-negative tumors.173 These lesions are notable for their aggressive histology and poor long-term outcome. This category overlaps but is not identical to the basaloid-like breast cancer category defined by microarray-based expression profiling174 and includes nonhereditary tumors that frequently express perturbations of BRCA1 pathway gene expression. The triple-negative category thus has increased the importance of accuracy in both positive and negative IHC testing and reemphasizes the need for local validation and controls for accurate IHC procedures (see the Best Practices section in this chapter). Finally, EGFR is expressed at a high level in most non–small cell lung carcinomas, including both adenocarcinoma and squamous carcinoma, and expression correlates with gene amplification and copy number.175,176 When a scoring system was applied in a recent phase 3 trial testing the effectiveness of the anti-EGFR chimeric (mouse/human) monoclonal antibody, Cetuximab, in advanced cases of non–small cell carcinoma, only patients with the highest levels of EGFR expression (scores >200) experienced a survival benefit.177 This result suggests that EGFR expression levels determined that IHC may be a reasonable selection criterion for anti-EGFR antibody treatment. Properly scored EGFR IHC expression may prove to be a valuable predictor of response to anti-EGFR monoclonal antibody treatment. The analysis of gene expression in cancer was accelerated greatly during the 1990s with the introduction of microarray technology that allowed simultaneous measurement of expression of large numbers of genes.178 The underlying technology of microarrays involves the binding of gene probes to specific addresses on a solid matrix. A fluorescence-labeled complementary DNA library is then hybridized to the solid matrix, and the level of expression at each address is measured by fluorescence microscopic scanning of the array. Many variations on this basic concept have resulted in high-density arrays that can be used to evaluate expression levels of virtually all genes in a single tissue sample. Large-scale gene expression profiling has been used to cluster tumors according similarities in gene expression patterns and to identify new subsets of lung and breast tumors,174,179–182 with prognostic implications. However, this method creates unwieldy amounts of data, and to produce a clinically relevant result, it has been necessary to filter expression data to identify profiles that can be used for clinical decision making. This refinement has been most thoroughly applied in breast cancer, where dominant competing platforms—Mammaprint (Agendia, Amsterdam, The Netherlands) and Oncotype DX (Genomic Health, Redwood City, Calif.)—have been created primarily for predicting likelihood of benefit from chemotherapy in early-stage breast cancer (reviewed in reference 183).183
Pathology, Biomarkers, and Molecular Diagnostics
Early Detection
Early Detection Research Network
Phase
Category
Descriptor
1
Preclinical/exploratory
Promising directions identified
2
Clinical assay and validation
Clinical assay detects established disease
3
Retrospective/longitudinal
Biomarker detects disease early, before it becomes clinical, and a screen positive rule is defined
4
Prospective screening
Extent and characteristics of disease detected by the test and the false referral rate are identified
5
Cancer control
Impact of screening on reducing the burden of disease on the population is quantified

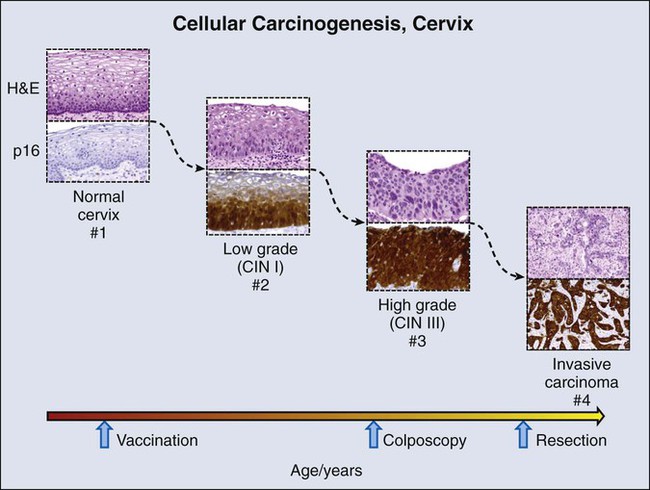
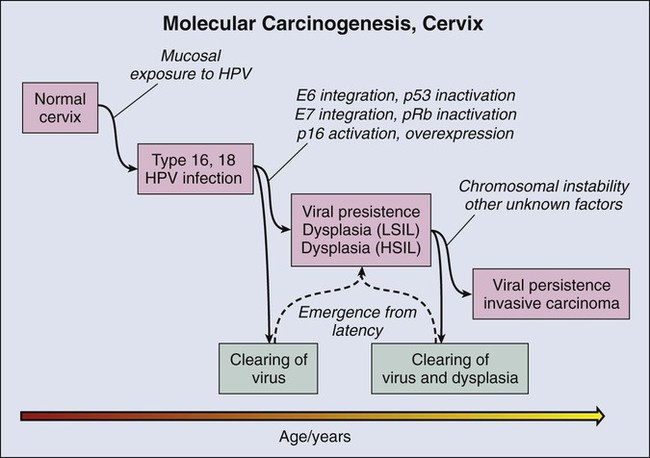
The Cervix and a Validated Biomarker
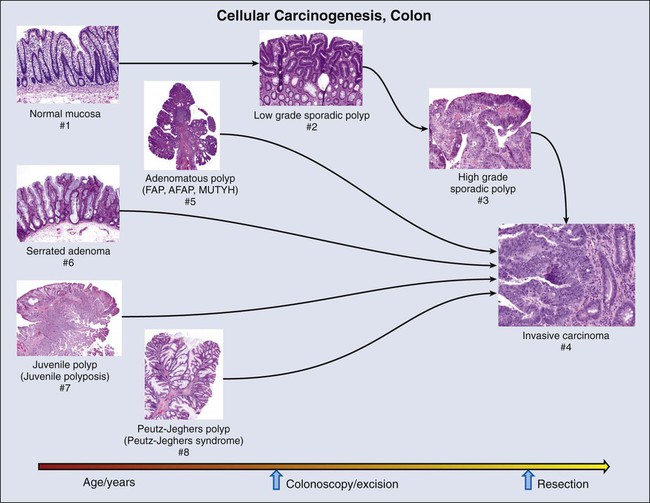
Colon and Multistep Carcinogenesis with Hereditary Components
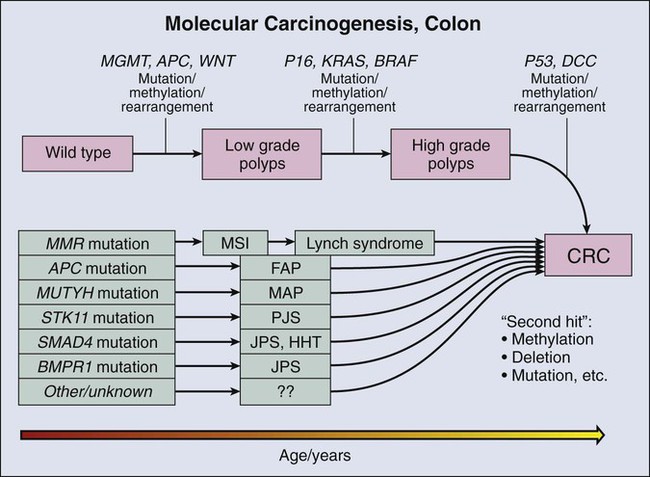
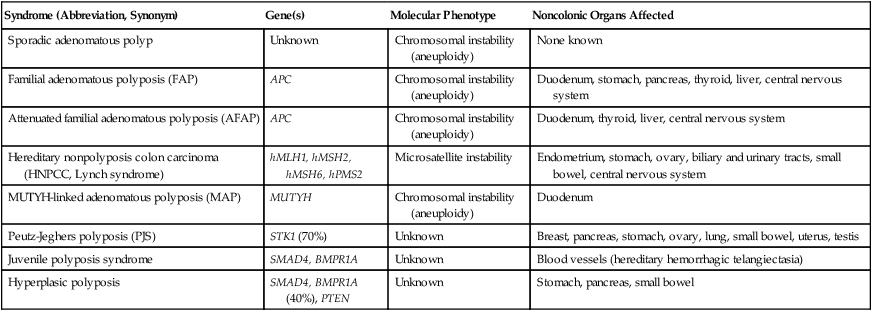
Syndrome (Abbreviation, Synonym)
Gene(s)
Molecular Phenotype
Noncolonic Organs Affected
Sporadic adenomatous polyp
Unknown
Chromosomal instability (aneuploidy)
None known
Familial adenomatous polyposis (FAP)
APC
Chromosomal instability (aneuploidy)
Duodenum, stomach, pancreas, thyroid, liver, central nervous system
Attenuated familial adenomatous polyposis (AFAP)
APC
Chromosomal instability (aneuploidy)
Duodenum, thyroid, liver, central nervous system
Hereditary nonpolyposis colon carcinoma (HNPCC, Lynch syndrome)
hMLH1, hMSH2, hMSH6, hPMS2
Microsatellite instability
Endometrium, stomach, ovary, biliary and urinary tracts, small bowel, central nervous system
MUTYH-linked adenomatous polyposis (MAP)
MUTYH
Chromosomal instability (aneuploidy)
Duodenum
Peutz-Jeghers polyposis (PJS)
STK1 (70%)
Unknown
Breast, pancreas, stomach, ovary, lung, small bowel, uterus, testis
Juvenile polyposis syndrome
SMAD4, BMPR1A
Unknown
Blood vessels (hereditary hemorrhagic telangiectasia)
Hyperplasic polyposis
SMAD4, BMPR1A (40%), PTEN
Unknown
Stomach, pancreas, small bowel
Familial Adenomatous Polyposis
Hereditary Nonpolyposis Colon Cancer
MUTYH-Associated Polyposis
Hamartomatous Polyposes
Juvenile Polyposis
Other Polyposes
Biomarkers for Colon Cancer Screening
Genetic tests
Other Screening Biomarkers
Lung Carcinogenesis and a Known Carcinogen
Premalignant Lesions in the Lung
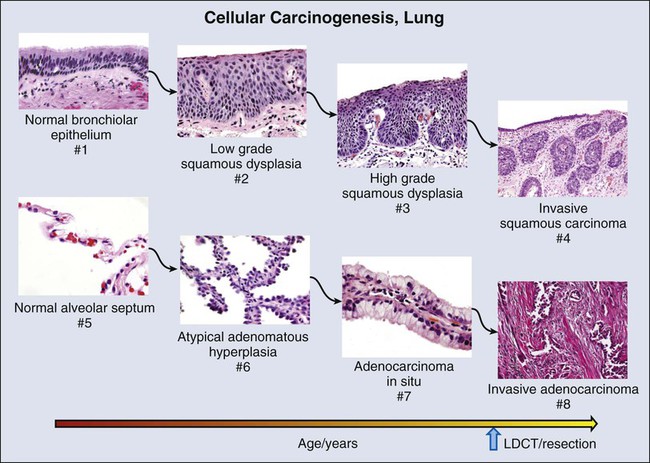
Squamous Dysplasia
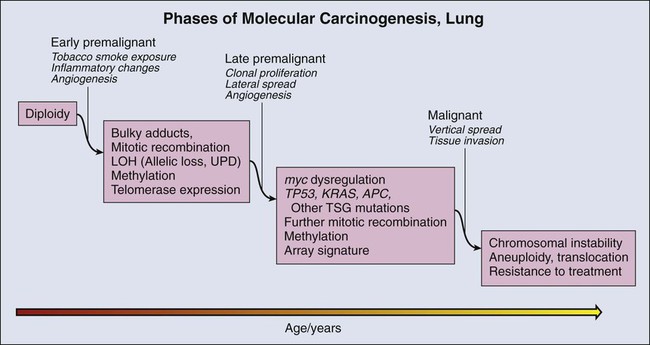
Molecular Changes during Lung Carcinogenesis
Tumor Suppressor Gene Methylation
Aneuploidy
High-Throughput Technologies
Other Organ Sites
Prognosis and Prediction
Protein Biomarkers for Prediction of Outcome
IHC Markers
Measurement of Gene Expression for Prognosis and Prediction of Treatment Response
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pathology, Biomarkers, and Molecular Diagnostics