Fig. 3.1
Conventional pancreatic ductal adenocarcinoma: variably sized well-formed glands surrounded by abundant desmoplastic stroma
The difficulty in distinguishing ductal adenocarcinoma from chronic pancreatitis also applies to the microscopic diagnosis and is regarded to be one of the most difficult distinctions in diagnostic pathology [6, 7]. Chronic pancreatitis may be associated with epithelial atypia, both architectural and cytologic, in pancreatic tissue; conversely, ductal adenocarcinoma is notorious for its deceptively bland appearance. Features favoring a malignant diagnosis include abnormal location of glands (adjacent to muscular arteries, within the duodenal muscularis, adjacent to adipocytes in the peripancreatic tissue, or around nerves), architectural abnormalities in the shape of the glands (cribriforming, angulation, or incomplete gland formation), and nuclear abnormalities (variation in the shape and size of nuclei by more than four to one, known as the 4-to-1 rule among the cells within an individual gland) [6, 7]. Diagnostic difficulty also extends to the differential diagnosis of ductal adenocarcinoma in metastatic sites, because ductal adenocarcinoma often retains its well-differentiated appearance and mimics benign or low-grade neoplasms of these sites. Common pitfalls include misinterpretation of metastatic ductal adenocarcinoma in the ovary as a primary borderline ovarian tumor [8]; in the lung, as bronchioloalveolar carcinoma; and in the liver, as bile duct adenoma. In the last instance, the converse – misinterpretation of bile duct adenoma as metastatic ductal adenocarcinoma – seems to be more common.
No uniformly applied pathologic grading system exists for ductal adenocarcinoma. Schemes advocated by Western and Asian experts show major philosophical differences in principle and results. The current American Joint Commission on Cancer-endorsed tumor-node-metastasis (TNM) grading system [9] is similar to the grading of other adenocarcinomas of the gastrointestinal tract: well differentiated means 95% or more composed of glandular structures; moderately differentiated, 50–95% glandular in pattern; and poorly differentiated, with more than 50% solid nest and individual cells. The World Health Organization (WHO) has adopted the complex grading scheme proposed by Klöppel and colleagues, which is difficult to employ and not widely used in daily practice [10]. Intratumoral heterogeneity seems to be an important problem in the grading of ductal adenocarcinoma, and for this reason, a simpler, more practical, and more clinically relevant grading scheme that accounts for this heterogeneity by scoring the patterns of infiltration has been proposed [11].
The pathologic evaluation of a pancreatectomy specimen is important both for staging and in determining the adequacy of resection. Recent studies have highlighted that, with more careful grossing protocols in pathology laboratories, in the vast majority of resected pancreatic ductal adenocarcinomas, there often are insidious carcinoma units that involve the surfaces and the margins which are not visible grossly or clinically [10–17]. In one study, in >90% of the cases, there were carcinomatous foci in the peripancreatic adipose surfaces of pancreatoduodenectomy specimens [18] which renders the current AJCC T-stage protocol inapplicable [9], and for this reason a size based staging protocol has been proposed [13, 19]. Metastasis to lymph nodes is considered one of the most important predictors of outcome in resected ductal adenocarcinomas. Generally, at least 12 lymph nodes should be identified in a simple pancreatoduodenectomy specimen [20]. Most of these lymph nodes are embedded in the surfaces of the pancreas or in the groove between the pancreas and duodenum. When careful harvesting of the lymph nodes is performed [12, 20], lymph node metastasis is detected in close to 80% of the resected pancreatic ductal adenocarcinomas [21].
Proper identification of margins and their adequate sampling are important components in the pathologic evaluation of a pancreatoduodenectomy specimen [12–17, 22]; however, what constitutes a margin remains controversial [12]. For example, the anterior surfaces are regarded as “margin” by some but not others. Similarly, whether to consider the posterior free surfaces of the pancreas as a “margin” or not has also been highly controversial, with vastly different views by different authors.
As expected, ductal adenocarcinoma shows immunohistochemical evidence of ductal differentiation. Briefly, most ductal adenocarcinomas express cytokeratins (7, 8, 18, 19, and variably 20), mucin (MUC1, MUC4, and MUC5AC), general adenocarcinoma markers (CEA, B72.3, CA125, and CA19-9), and some pancreatic cancer-specific markers (mesothelin, S-100A4, etc.) [23–25]. In addition, immunolabeling for antigens that function as surrogate markers for genetic changes can also be altered in ductal adenocarcinomas. Most ductal adenocarcinomas have abnormal nuclear labeling with antibodies to p53, and 55% show a loss of SMAD4 expression [26].
The genomes or exomes of a large number of ductal adenocarcinomas have been sequenced, significantly increasing our understanding of the molecular drivers of pancreatic cancer. Although the genetic changes identified are complex, the key to understanding pancreatic tumorigenesis lies in the recognition and appreciation that these mutations target a core set of pathways and processes. Mutation in codon 12 of the KRAS oncogene is found in more than 95% of ductal adenocarcinoma and seems to be an early event [27]. Mutation of P16 or methylation of the promoter is common (>80%) and represents the pathogenetic link with the familial atypical multiple mole melanoma syndrome [28] and thus have clinical implications for patient and family screening. Overexpression of p53 [29, 30] and loss of SMAD4/DPC4 are detected in about half of cases [26]. BRCA2 and Peutz–Jeghers gene mutations have been implicated in about 5% of ductal adenocarcinoma cases [27]. BRCA has been the subject of much discussion recently, because of the potential targeting agents in the treatment of such cases [31]. Fanconi anemia gene alterations also have been identified [32]. Abnormalities in mismatch repair proteins and microsatellite instability are uncommon, although pancreatic ductal adenocarcinomas can occur as one of the less common manifestations of Lynch syndrome [33].
3.1.2 Pancreatic Intraepithelial Neoplasia (PanIN)
The vast majority of pancreatic ductal adenocarcinomas are believed to arise from precursor intraductal proliferations termed pancreatic intraepithelial neoplasia (PanIN) [34]. PanINs are small microscopic intraductal lesions that are less than 5 mm in size. They are composed of a flat or papillary neoplastic epithelium. The spectrum of changes, originally classified in three grades [35], is now being modified into a two-tier system as low versus high-grade PanIN [36]. Replacement of the normal cuboidal, nonmucinous ductal epithelium with columnar cells that contain abundant apical mucin, but without architectural complexity (e.g., papilla formation) or cytologic atypia (previously called mucinous metaplasia, mucinous hypertrophy, and more recently, PanIN-1A), is regarded as the earliest form of neoplastic transformation in the pancreatic ductal system (Fig. 3.2a). As the intraductal neoplasm progresses, it acquires more papillary architecture and cytologic atypia. When irregular papillary architecture is present with tufting, severe cytologic atypia, necrosis, suprabasal mitoses, and loss of cell polarity, it is regarded as high-grade PanIN (previously called PanIN3; high-grade dysplasia), which is equivalent to “carcinoma in situ” (Fig. 3.2b).
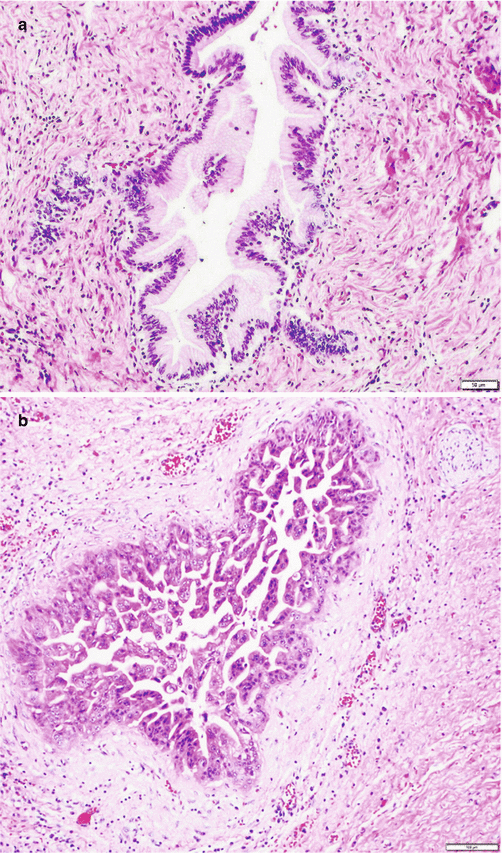
Fig. 3.2
(a) Pancreatic intraepithelial neoplasia, low grade; (b) pancreatic intraepithelial neoplasia, high grade
A progressive accumulation of molecular alterations is reported from low-grade PanIN to invasive carcinoma [27]. Some alterations, such as KRAS mutations, are early events; while others, such as p53 overexpression, occur at the more advanced end of this spectrum. Low-grade PanINs are very common incidental findings in the normal population [37, 38]; therefore, they are generally believed to not to require any further clinical attention, if encountered in isolation or at resection margins. In fact, it is not required to even record it in the surgical pathology report [36]. High-grade PanIN (previously PanIN3/CIS), on the other hand, is seldom seen in the absence of an invasive carcinoma [37], and for this reason, if high-grade PanIN (CIS) is encountered in a pancreas, the likelihood of carcinoma elsewhere in the gland is very high. In fact, one of the biggest challenges is to define and distinguish high-grade PanINs from colonization (cancerization: intraductal spread of invasive carcinoma), i.e., invasive carcinoma cells that retrogradely infiltrate into the native ducts and “colonize” them and grow “pagetoidly” within the duct epithelium (cancerization), versus true precursor.
3.1.3 Other Carcinomas of Ductal Origin
There are other malignant neoplasms of ductal origin/lineage (i.e., carcinoma types) that may be related to ductal adenocarcinoma (and some also associated with ordinary ductal adenocarcinoma component) but are classified separately because of their distinctive clinical and molecular characteristics. In the following section, the salient features of these tumor types are discussed.
Colloid carcinoma is characterized by the production of copious amounts of extracellular mucin [39–41] (Fig. 3.3), and these distinctive neoplasms almost always arise in association with an intestinal-type intraductal papillary mucinous neoplasm (IPMN). The majority of intestinal-type IPMNs (as well as their associated carcinomas) harbors GNAS gene mutations, and, of interest, colloid carcinomas have a different biology with an unusually protracted clinical course [24, 42]. Overall, they have an incomparably better prognosis than ductal adenocarcinomas, with 5-year survival of >55% [39–41]. Since colloid carcinomas arise mostly from “intestinal-type” IPMNs, and since both show diffuse expression of intestinal lineage markers of CDX2/MUC2 which are otherwise practically nonexistent in other tumor types of this organ, it is being speculated that colloid carcinomas may have to be managed like an intestinal-type cancer (with the intestinal chemotherapy protocols) rather than pancreatic.
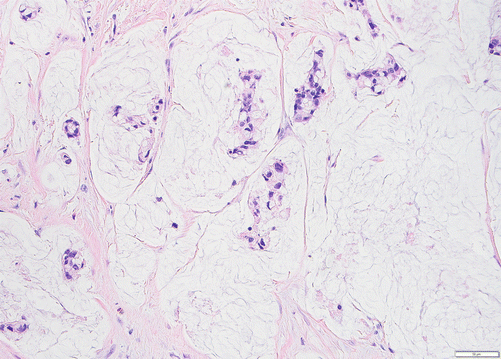
Fig. 3.3
Colloid carcinoma is a distinct indolent form of adenocarcinoma characterized by mucin lakes with malignant glands floating within. It is speculated that protracted clinical course of this type of carcinoma is attributable to the containing effect of the mucin
The medullary variant of pancreatic cancer is a poorly differentiated pancreatic cancer (Fig. 3.4). Syncytial nodules of large, poorly differentiated epithelial cells with a pushing pattern of invasion characterize medullary carcinomas. In our experience, these are significantly more common in the ampulla and duodenum than in the pancreas, and therefore, before a case can be classified as a pancreatic origin, these possibilities ought to be excluded. In fact, most of the cases advertised as pancreatic medullary carcinoma prove to be of ampullary origin in careful inspection. Medullary carcinoma can occur sporadically or in patients with Lynch syndrome [43]. Many but not all tumors are microsatellite instable, and immunostaining for mismatch repair proteins is lost in some of these cancers [44, 45]. The diagnosis of medullary carcinoma of the pancreas may be a clue to an inherited cancer syndrome, including Lynch syndrome, and may justify genetic counseling of the patient. In one study [44], these tumors were found to have a more protracted clinical course, but further data are necessary to define the prognosis of these rare tumors.
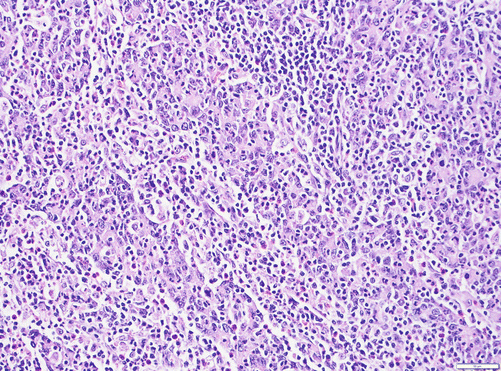
Fig. 3.4
Medullary carcinoma is a poorly differentiated carcinoma that mostly lacks gland formation and is characterized by syncytial growth of large atypical cells accompanied by a lymphocytic infiltrate
Undifferentiated carcinoma can be regarded as the least differentiated form of ductal adenocarcinoma, in which characteristic tubule formation is no longer evident or only focal. This term is unfortunately applied for ordinary ductal adenocarcinomas with significant nonglandular (poorly differentiated) component. Defined more stringently, undifferentiated carcinomas appear to occur in two different types. One is characterized by rhabdoid phenotype and common INI-1 loss, which is very uncommon [46], and the other is osteoclastic giant cell carcinoma [47]. The latter undifferentiated carcinoma with osteoclast-like giant cells (also known as osteoclastic giant cell carcinoma) is a distinctive tumor characterized by an abundance of osteoclasts in the background of a sarcomatoid carcinoma [47–49] (Fig. 3.5). Studies have shown that the osteoclastic giant cells are nonneoplastic histiocytic cells [49], and the true neoplastic cells in this tumor are the sarcomatoid mononuclear cells. An adenocarcinoma component or, in some cases, high-grade PanIN or mucinous cystic neoplasm precursors may be present. Undifferentiated carcinomas with osteoclast-like giant cells are characterized by a well demarcated and a large solitary mass and exhibit nodular/pushing-border infiltration [47]. If examined carefully, many such tumors appear to have substantial intraductal growth. These are clearly malignant neoplasms; however, careful reappraisal of the literature elucidates that their prognosis is significantly better than that of ordinary ductal adenocaricnomas. In fact, in our experience, many of these patients experience unexpectedly long survival, with an overall 5 years of 42% [47], but more studies are needed to verify this impression.
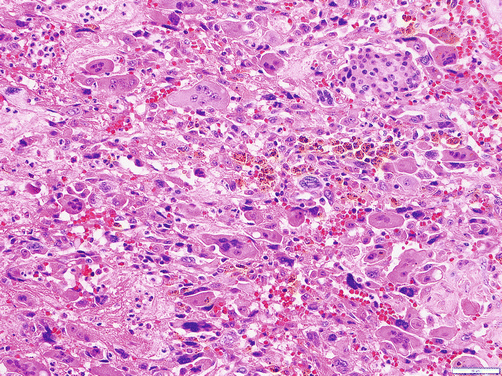
Fig. 3.5
Undifferentiated carcinoma with osteoclast-like giant cells: it is characterized by osteoclasts (benign multinucleated giant cells of osteoclastic type) admixed with pleomorphic sarcomatoid carcinoma cells and often accompanied by hemorrhage and hemosiderin in the tumor nodules
Squamous differentiation is seen in some conventional ductal adenocarcinomas (i.e., adenosquamous carcinomas (Fig. 3.6), but rare pure examples of squamous cell carcinoma without any glandular components also may be seen [50] though exceedingly uncommon. They may have variable degrees of keratinization. Squamous cell carcinoma and adenosquamous carcinoma of this region are highly aggressive tumors [50] with a prognosis that is even worse than that of ordinary ductal adenocarcinoma.
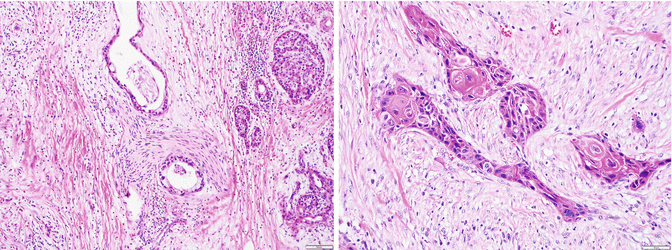
Fig. 3.6
Adenosquamous carcinoma: left, invasive adenocarcinoma component; right, large nests of cells with squamous differentiation including keratinization (pearl formation)
3.1.4 Intraductal Neoplasms
Intraductal neoplasms are the generic category designation for the tumors that fundamentally arise in the main pancreatic duct or its branches. There are three types of pancreatic neoplasms that predominantly have an intraductal growth pattern: the common, usually cystic, intraductal papillary mucinous neoplasms; the rare, usually solid intraductal tubulopapillary neoplasms; and the rare “intraductal tubular pyloric gland-type adenoma,” which is mostly regarded as a subset of one of the former entities [51]. In addition to these three tumor types, pancreatic neoplasms with a usually solid growth pattern such as acinar cell carcinomas [52, 53], neuroendocrine tumors, metastatic tumors, and undifferentiated carcinomas [47] may present, though very rarely, as predominantly intraductally growing neoplasms.
3.1.4.1 Intraductal Papillary Mucinous Neoplasms
Intraductal papillary mucinous neoplasms (IPMNs) account for at least 25–30% of all neoplastic cystic lesions [3]. By definition, IPMNs are mucin-producing epithelial neoplasms that involve the duct system and are equal to or larger than 1 cm in size. These neoplasms are noninvasive and can harbor varying degrees of dysplasia. Most arise in the head of the pancreas; however, they may also arise in the tail, and some even involve the entire pancreas [54–56]. The mucin produced by these tumors may exude from the ampulla of Vater, a finding that is virtually diagnostic of an IPMN. Radiographic findings of ductal dilatation with irregularities are often diagnostic as well. IPMNs may be multicentric, and therefore the presence of one lesion should heighten the clinical suspicion for additional lesions and mandate careful follow-up. Macroscopically, IPMNs are characterized by dilatation of the main or branch pancreatic ducts. Papillary fronds of neoplastic epithelium and tenacious luminal mucin are often present. Microscopically, the neoplastic epithelium can be papillary or flat and can show one of four directions of differentiation: intestinal, gastric, pancreatobiliary, or oncocytic [57–59]. In intestinal-type IPMNs, the papillary nodules are morphologically identical to colonic villous adenomas (Fig. 3.7a), and the invasive carcinomas that develop in these tend to be of the relatively indolent colloid type [39]. Intestinal-type IPMNs and colloid carcinomas typically express intestinal differentiation markers (MUC2 and CDX2) not found in ductal adenocarcinomas or in the nonintestinal subtypes of IPMNs discussed below, indicating that they represent a distinct “intestinal pathway” of carcinogenesis in the pancreas [57]. This is highly pertinent to the management of these tumors, because colloid carcinomas not only behave in a much more protracted clinical course but also may be closer in biology to the intestinal than pancreatic adenocarcinomas and may have to be treated as such. The pancreatobiliary-type IPMNs, which are least well characterized, typically have complex arborizing and interconnecting papillary configuration with delicate fibrovascular cores and are composed of cuboidal cells with enlarged nuclei and little mucin production (Fig. 3.7b). This subtype tends to be associated with tubular-type invasive carcinoma (conventional ductal adenocarcinoma) and appears to have more aggressive behavior [60]. The gastric-type IPMNs are the most common type since most “incidentaloma cysts” of the pancreas prove to be this group. It is characterized by relatively simple and typically short papillae and often has pyloric-like glandular elements at their base in the cyst wall. The epithelial lining is identical to gastric foveolar epithelium (Fig. 3.7c). When high-grade dysplasia ensues on gastric-type IPMN, it typically starts to show more complex architecture and cuboidal cells with enlarged nuclei and less mucinous cytoplasm, which are also characteristics of the pancreatobiliary-type IPMN mentioned above. For this reason, some authors believe the pancreatobiliary type is a high-grade version of the gastric type [51]. What is currently classified as oncocytic type IPMN, which had been originally described as intraductal oncocytic papillary neoplasm, is now proving to be different not only morphologically but also by clinical behavior from other IPMN types and thus deserves to be recognized as a distinct entity [59, 61–63]. This entity is characterized not only by the oncocytic nature of the cells but also by the complexity of the papillary nodules, which have an arborizing pattern (Fig. 3.7d). Oncocytic IPMNs are often large and very floridly proliferative tumors and clinically they typically get diagnosed as “cancers” with cystic component [63]; whereas, the carcinomas arising from them tend to be small, the incidence of metastasis is very low, and the overall prognosis appears to be very favorable [59, 61, 62]. In fact, despite their large size and complexity, very little mortality has been attributed to this tumor type, if any. On the other hand, they also have a tendency to recur. Overall, clinicopathologic and behavioral characteristics of these oncocytic lesions are so distinctive and different from other IPMNs that they need to be regarded as a separate category [59, 61–63]. Recent molecular studies demonstrate that GNAS mutations are more prevalent in intestinal compared with pancreatobiliary and gastric subtypes [64–66] and oncocytic IPMNs have different molecular changes from other IPMNs, with a much lower incidence of KRAS mutation and frequent expression of MUC6, suggesting a pyloropancreatic lineage [23, 62].
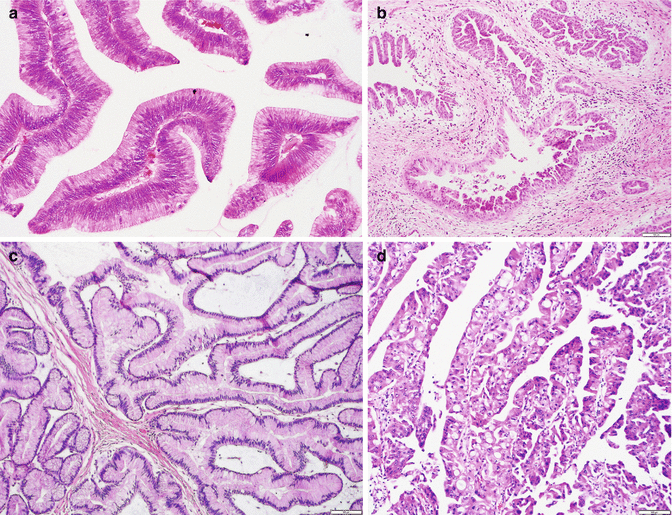
Fig. 3.7
Intraductal papillary mucinous neoplasm: (a) intestinal type resembling colonic villous adenoma; (b) pancreatobiliary type (with cuboidal cells and round nuclei); (c) gastric type (identical to gastric foveolar epithelium); (d) intraductal oncocytic papillary neoplasm (aka oncocytic variant of IPMN showing markedly complex papillae forming cribriform and/or solid areas; the tumor cells exhibit intracellular lumina and contain abundant eosinophilic granular cytoplasm and single prominent eccentric nucleoli)
Thus, these different histologic subtypes of IPMNs not only have different progression rates and different associations with biologically distinct invasive carcinoma types, but also are representing distinct pathways of carcinogenesis. Although IPMNs are typically classified into a histological subtype, more than one epithelial subtype can be present within the same IPMN. The degree of dysplasia in IPMNs is now grade in a two-tiered system as low grade (encompassing the previous low- and intermediate-grade dysplasia categories) versus high grade (which is reserved for the cases that used to be qualified as “carcinoma in situ” [36, 55]. Among patients with IPMNs who go to pancreatic resection, about 30% will have IPMNs that have an associated invasive adenocarcinoma [51] although this figure may be changing with earlier detection and better selection of cases for surgery. Patients with an invasive carcinoma arising in an IPMN have a better prognosis than do patients with a conventional ductal adenocarcinoma not arising in an IPMN, but some of this improved prognosis is lost when one controls for stage [51]. Invasive carcinomas that arise in IPMNs are recognized separately and are graded and staged like other ductal-type carcinomas [36, 55].
3.1.4.2 Intraductal Tubulopapillary Neoplasms
Intraductal tubulopapillary neoplasm (ITPN) [56, 57, 67–69] is a recently recognized category of mass-forming (>1.0 cm) intraductal neoplasm that is fairly similar to an IPMN, from which it is distinguished microscopically by its mucin-poor nature and distinctive tubular architecture. First reported by Tajiri and colleagues [67] under the heading of intraductal tubular adenocarcinoma, the entity is now being designated intraductal tubulopapillary neoplasm in the WHO 2010 classification. It is a rare tumor seen at an average age of 53 years, and it presents with nonspecific symptoms [56]. The clinical findings are similar to those of IPMNs but generally forming more complex nodular tumors on imaging as well as macroscopic examination [56]. Cystic change is often less appreciable. ITPN occurs predominantly in the head of the pancreas but may involve any part. It is often large (mean, 7 cm; range, ≤15 cm) [56].
Histologically, ITPNs are characterized by densely packed cuboidal eosinophilic epithelial cells forming intraductal tubular proliferations, usually with moderate nuclear atypia, increased mitotic activity, and without overt mucus production (Fig. 3.8). Occasionally the glands form a tubulopapillary pattern and some cases have comedo-like necrosis or desmoplastic stroma. The tumor cells are positive for the ductal CKs [7, 8, 18, 19] and, in more than 60% of cases, for MUC1 and MUC6. MUC5AC and MUC2 are negative [56]. In about 40%, there is an invasive carcinoma component, which may be difficult to recognize but is usually limited in extent [56]. The prognosis is significantly better than that of PDACs. More than a third of the patients survive beyond 5 years, and a protracted clinical course may be even seen in those patients with recurrence and metastasis to lymph nodes or to the liver [56]. Molecular pathways involved in this tumor appear to be very different than those of ordinary ductal adenocarcinomas or IPMNs [70].
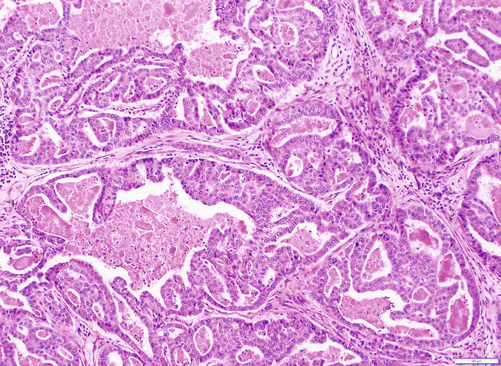
Fig. 3.8
Intraductal tubulopapillary neoplasm: it is composed of irregular tubules forming a large and cribriform mass
3.1.5 Mucinous Cystic Neoplasms
Mucinous cystic neoplasms (MCNs) of the pancreas are cystic mucin-producing neoplasms. Now defined by the presence of ovarian-type stroma, this entity has highly distinctive characteristics. The vast majority occur in perimenopausal women (98% female; mean age, 48 years) [71–73] with only few male patients with convincing ovarian stroma on record. Most (>98%) occur in the body/tail; they are very uncommon in the head. They form a relatively distinct (demarcated) lesion. In contrast to IPMNs, the cysts of MCN do not communicate with the larger pancreatic ducts. The cysts are most frequently multiloculated and distended with tenacious mucin, which is rich in glycoproteins and oncoproteins, such as CEA [74–77]. This feature may help distinguish these tumors from other cystic lesions. Grossly, the inner surfaces of the cyst walls may be smooth, they may have papillary excrescences, or they may have isolated intracystic solid nodules. Microscopic examination shows two characteristic components: variable lining, from low-cuboidal/nonmucinous to tall-columnar/mucinous arranged in a flat or papillary architecture, and distinctive ovarian-type stroma, the cells of which may express estrogen receptors [4] and also some progesterone receptors (Fig. 3.9). Based on the degree of cytoarchitectural abnormalities on the most atypical region, these neoplasms are now graded into two groups: low grade (previously called low- or intermediate-grade dysplasia) or high grade (previously called high-grade dysplasia and also corresponding to “in situ carcinoma”) [36]. Invasive carcinoma is seen in about 15% of the cases, typically in larger and more complex examples that show florid papillary nodules in the cysts; invasion is seldom seen in tumors that are small (<3 cm) and noncomplex [73], raising the question of whether these may be amenable for watchful waiting in select patients as in IPMNs. Most invasive carcinomas are tubular (ductal) type and morphologically indistinguishable from ordinary ductal adenocarcinomas. A few are sarcomatoid carcinomas, some with osteoclastic giant cells [47, 73]. Recent literature indicates that if invasion has been ruled out by total sampling and thorough examination of the tumor, then noninvasive MCNs behave in a benign fashion [72, 73]. In contrast, those with invasive carcinoma appear to exhibit fairly aggressive clinical course, even when they are small. Having said that, in one recent study, those with “minimal invasion,” defined as carcinoma limited to microscopic foci within the septa of the cysts, were found to have a fairly benevolent behavior [78]. Recently, the exomes of a series of well-characterized MCNs have been sequenced, and the KRAS, p16, p53, RNF43, and SMAD4 genes have been reported to be targeted in MCNs [4].
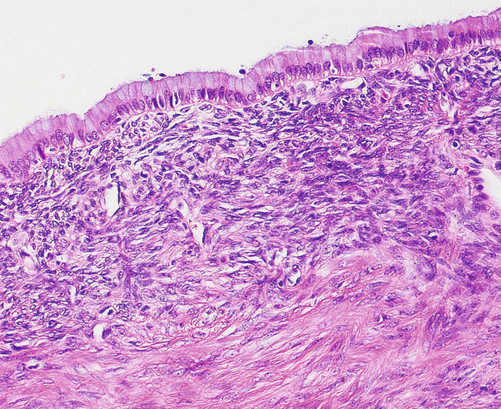
Fig. 3.9
Mucinous cystic neoplasm: tall-columnar mucinous epithelium is surrounded by an ovarian-type stroma
3.1.6 Serous Cystic Tumors
Serous cystadenomas are rare benign neoplasms that can form relatively large masses (up to 25 cm) that tend to be well demarcated, predominantly in women and in the age group of 50s–60s [79]. Typically, they are composed of innumerable back-to-back tubules of variable size and shape creating the diagnostic macroscopic appearance of a spongelike configuration – characteristic microcystic adenoma (Fig. 3.10). Rare macrocystic (oligocystic), unicystic, and solid examples occur, and recent studies have shown that these do not significantly differ from the more common microcystic variant perhaps with the exception of their higher propensity to mimic (and be misdiagnosed as) other megacystic tumors or neuroendocrine neoplasms [79]. Microcystic serous cystadenomas often have a central stellate scar. Microscopically, they are characterized by distinctive glycogen-rich epithelial cells with uniform round nuclei; dense, homogenous chromatin; and a prominent epithelium-associated microvascular meshwork [80] (Fig. 3.10). Serous cystic tumors are one of the few ductal neoplasms of the pancreas that do not produce mucin, possibly reflecting a recapitulation of the centroacinar cells that are nonmucinous. Along the same lines, the cyst contents are devoid of the mucin-related glycoproteins and oncoproteins that typically are found in mucinous pancreatic tumors, a feature that may help in the preoperative diagnosis [81]. Instead, they appear to produce a fair amount of vascular endothelial growth factor (VEGF) and secrete it to the cyst fluid, which may be helpful in the preoperative diagnosis [80, 82]. Microscopically, these lesions are similar to the cysts seen in von Hippel–Lindau (VHL) disease, and some serous cystadenomas do show VHL gene alterations. Serous cystadenomas often are reported to coexist or “collide” with other pancreatic neoplasms (especially neuroendocrine neoplasms) and with congenital pathologic conditions [79, 82].
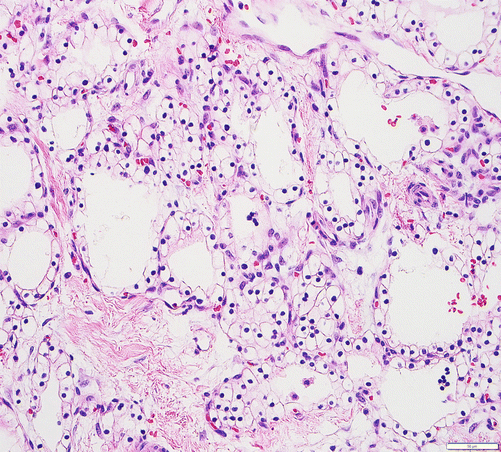
Fig. 3.10
Microcystic serous cystadenoma: each small cyst is lined by a flattened layer of epithelium; cytologically, the lining cells show clear cytoplasm and small, uniform, hyperchromatic nuclei. Intimately intermixed with the epithelium is a continuous layer of capillary-sized vessels
Convincing examples of malignant serous tumors (serous cystadenocarcinomas or carcinomas-ex-microcystic adenoma) are exceedingly rare [83] and are in fact dubious as to whether they really represent a malignant counterpart of serous neoplasm. Most of the cases reported in the literature as “malignant” serous cystic neoplasm (SCN) however appear not to qualify for the current WHO definition of malignancy for these tumors [79]. For example, SCNs that radiologically about the large vessels have been designated as “malignant.” Also, larger SCNs can show adhesion to the neighboring organs [79, 84] such as lymph nodes, spleen, stomach, and colon, which may not necessarily be a sign of true malignant behavior. Similarly occasional recurrences exhibited by SCNs may be a simple persistence of tumor that can be seen in some benign neoplasms, rather than true malignant behavior. Even the cases with synchronous liver involvement may in fact represent simultaneous independent involvement similar to what is seen in VHL cases. There has not been any documentation of metastatic SCN in distant organs other than the liver-involving cases (many of which may merely be synchronous disease) and there has not been any example with widely disseminated disease. We are aware of cases in the liver that were designated as “serous cystadenocarcinoma” who are alive without disease many years after the resection. And in critical review of the literature, most such cases appear to be like that. Thus, for practical purposes, serous cystadenomas limited to the pancreas are regarded as uniformly benign [79].
3.1.7 Pancreatic Neuroendocrine Tumors
Pancreatic neuroendocrine tumors (PanNETs) are the second most common malignancy of the pancreas [3]. Most neuroendocrine-type tumors of the pancreas are well-differentiated (low- to intermediate-grade) neuroendocrine tumors, previously referred to as islet cell tumors/carcinomas and now designated as pancreatic neuroendocrine tumors (PanNETs) by the 2010 WHO [85, 86], with low-grade malignant behavior [87]. Grossly, PanNETs are usually solid, circumscribed, and fleshy tumors, although multinodular and sclerotic examples occur [85, 86]. Rarely, cystic degeneration may be seen [75, 88–90], with a central unilocular cyst lined by a cuff of viable tumor; this occurs more often in the setting of multiple endocrine neoplasia (MEN) syndrome type I. These cystic examples may be more benevolent [75, 88–90]. PanNETs recapitulate the morphologic features of islet cells by forming nested, gyriform, trabecular, or rarely acinar or glandular patterns (Fig. 3.11). The cells have characteristic neuroendocrine features, including round, monotonous nuclei with a coarsely stippled chromatin pattern and moderate amounts of cytoplasm (Fig. 3.11).
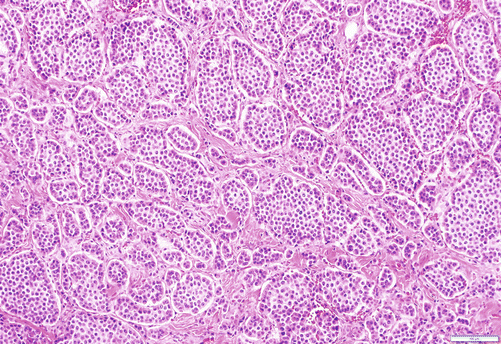
Fig. 3.11
Pancreatic well-differentiated neuroendocrine tumor: it shows nests and/or trabecular pattern of relatively round uniform epithelial cells with a fair amount of cytoplasm and salt-and-pepper chromatin
Almost half of PanNETs are clinically functional, and functional PanNETs can be further subclassified based on the clinical syndrome they produce (not based on immunohistochemical hormone expression). The most common functional PanNETs are insulinomas, while gastrinomas, glucagonomas, somatostatinomas, and VIPomas (vasoactive intestinal peptide tumor) are rarer. Most insulinomas follow a benign clinical course, likely because insulinomas typically are highly symptomatic, even when they are small, which leads to their early detection. Glucagonomas, on the other hand, tend to be large at diagnosis and have a more aggressive course. Nonfunctional PanNETs constitute an ever-enlarging proportion of PanNETs because they are being detected more commonly as incidental findings on abdominal imaging studies [91, 92]. PanNETs associated with MEN type I or other syndromes tend to be multifocal and less aggressive [93–95]. In addition to grossly evident and usually functional PanNETs, patients with MEN type I have numerous neuroendocrine microadenomas, defined as PanNETs (<0.5 cm). Most sporadically occurring functional and nonfunctional PanNETs are clinically low-grade malignancies. More than half of patients have recurrence or metastasis after resection, and many patients come to attention only after the development of metastatic disease. Nonetheless, there may be a relatively protracted clinical course even in patients with metastatic disease. It has been difficult, as in neuroendocrine tumors of other organs, to determine which PanNETs are more likely to metastasize and which metastatic cases are likely to progress most rapidly [96, 97]. Findings associated with more aggressive behavior include a size greater than 3 cm, a functional PanNET other than insulinoma, extrapancreatic or vascular invasion, high mitotic activity, high proliferation index (based on immunohistochemical staining for Ki-67) [98–100], CK19 expression [101], and c-KIT expression [102]; however, some PanNETs lacking all of these features may still metastasize. The genes targeted in neuroendocrine tumors, which differ significantly from those targeted in ductal adenocarcinomas, include MEN1, DAXX and ATRX, and genes coding for members of the mammalian target of rapamycin (mTOR) pathway [4].
Recently, a multidisciplinary group of international experts have proposed a set of parameters to be included in pathology reports [103]. It was emphasized that PanNETs ought to be evaluated by the approach used for any other malignancy, and accordingly, the grade and stage should be reported separately [85, 104]. For grading, the 2010 WHO adopted the system originally devised and tested by the European Neuroendocrine Tumor Society (ENETS), which grades PanNETs based on the mitotic count and Ki-67 labeling index. It is grade 1 if the mitotic rate is 0–1/10 HPFs or the Ki-67 index is below 3%, grade 2 if the mitotic rate is 2–20/10 HPFs or the Ki-67 index is 3–20%, and grade 3 if either is above 20. Recent studies have shown that the G3 category actually includes at least two different tumor types with different morphological, genetic, and clinical features: histologically uniform NETs with an elevated proliferative rate and poorly differentiated NEC with small cell or large cell morphology [4, 105, 106]. For staging, the AJCC has adapted the TNM-based staging system used for adenocarcinomas, but the ENETS system is somewhat different [107].
3.1.8 Acinar Neoplasms
Although acinar tissue constitutes most of the pancreas, acinar neoplasms are rare, most being acinar cell carcinomas. Acinar neoplasms are characterized by the production of pancreatic enzymes, such as trypsin, chymotrypsin, and lipase. Solid acinar cell neoplasms are carcinomas; although a benign cystic variant exists, known as acinar cell cystadenoma, no solid acinar cell adenoma has been defined in the pancreas.
Acinar cell carcinomas form relatively large tumors (mean, 10 cm), usually in older men (mean age, 63 years) [108–110], although some occur in children [111]. In a small percentage of cases (10%), patients experience a “lipase hypersecretion syndrome” [112] characterized by subcutaneous fat necrosis, polyarthralgia, and peripheral eosinophilia. Serum α-fetoprotein (AFP) levels may be elevated in some cases [113]. Half of acinar cell carcinomas have metastases at the time of diagnosis, usually in the liver and/or regional lymph nodes [114]. Once believed to be almost as aggressive clinically as ductal adenocarcinomas with a 5-year survival of 10% [114], more recent studies place acinar cell carcinomas in a somewhat more indolent category [115] with some studies reporting a 5-year survival over 40% [108, 110]. In contrast to ductal adenocarcinomas, acinar cell carcinomas are stroma-poor cellular neoplasms with acinar cell differentiation, based on morphology and immunohistochemical staining for acinar enzymes, especially trypsin and chymotrypsin [115]. The cyanophilic acinar-appearing cells typically exhibit granular cytoplasm and centrally located nucleus with a prominent nucleolus. The cells are arranged in sheets and trabecular pattern [116] (Fig. 3.12). A subset of acinar cell carcinomas is characterized by prominent intraductal growth; such cases appear to be associated with a more protracted clinical course [52, 53]. Molecular genetic findings of acinar cell carcinomas markedly differ from those of ductal adenocarcinomas [117], with absence of the common alterations of ductal adenocarcinoma in genes such as KRAS, TP53, P16, or SMAD4.
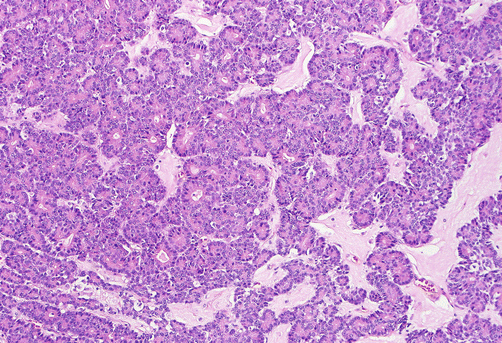
Fig. 3.12
Acinar cell carcinoma: it commonly exhibits a solid growth pattern, with sheets and nests of cells with moderate amount of amphophilic cytoplasm and minimal lumen formation
Immunohistochemistry discloses scattered neuroendocrine cells in 30–40% of acinar cell carcinomas. Some cases have a significant neuroendocrine component that may be evident microscopically [115]. If the latter constitutes more than 25% of the tumor, it is classified as mixed acinar-neuroendocrine carcinoma, a tumor that seems to be biologically similar to pure acinar cell carcinoma [118, 119]. Similarly, acinar cell carcinomas with more than 25% ductal differentiation are classified as mixed acinar-ductal carcinoma [120]. Rarely, acinar cell carcinomas may show a grossly cystic pattern and are designated acinar cell cystadenocarcinoma [121, 122].
Acinar cell cystadenoma, also referred to as acinar cystic transformation, is a rare entity [123, 124]. They are usually small, incidental cysts lined by benign-appearing acinar cells, although some may form a mass that measures a few centimeters. The cysts may be patchily distributed amidst pancreatic parenchyma. Typically, the lining cells are often nondescript and intermixed with other cell types including nonmucinous ductal-type cells. In fact, acinar cells may be a relatively smaller population in the process. Some cases show nodular growth of proliferating acinar cells [125]. They are benign, non-clonal processes [125].
3.1.9 Solid Pseudopapillary Neoplasm
Solid-pseudopapillary neoplasms (SPNs) are rare solid neoplasms of the pancreas and typically occur in young women (mean age, 25 years; >80% female) [126–129] although they can be encountered at any age and in men as well. It is a peculiar neoplasm of unknown origin, and its obscure nature is reflected in the various descriptive names assigned to this tumor in the past, including papillary cystic tumor, solid and papillary tumor, solid and cystic tumor, and Frantz tumor [130–133]. It is a very indolent “malignant” neoplasm for which complete resection is curative in most cases. Metastases are very uncommon, usually to liver or peritoneum and often at the time of diagnosis [129, 134]. Even patients with metastases appear to have a protracted clinical course, and death as a result of this tumor is rare [134]; however, very rare examples of high-grade sarcomatoid malignant transformation form a conventional solid pseudopapillary neoplasm [129].
Related posts:
 Staging and Determination of Resectability of Pancreatic Cancer
Laparoscopic Distal Pancreatectomy in Pancreatic Cancer
Staging and Determination of Resectability of Pancreatic Cancer
Laparoscopic Distal Pancreatectomy in Pancreatic Cancer
 PET and Other Functional Imaging
PET and Other Functional Imaging
 Mesenteric Approach for Pancreaticoduodenectomy
Mesenteric Approach for Pancreaticoduodenectomy
 Role of Extended Resection in Pancreatic Cancer
Role of Extended Resection in Pancreatic Cancer
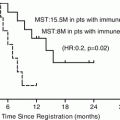 Emerging New Treatment Modalities: Irreversible Electroporation
Emerging New Treatment Modalities: Irreversible Electroporation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


