The pathologic classification of neuroendocrine neoplasms has evolved over the past decades, as new understanding of the biological behavior, histologic characteristics, and genetic features have emerged. Nonetheless, many aspects of the classification systems remain confusing or controversial. Despite these difficulties, much progress has been made in determining the features predicting behavior. Genetic findings have helped establish relationships among different types of neuroendocrine neoplasms and revealed potential therapeutic targets. This review summarizes the current approach to the diagnosis, classification, grading, and therapeutic stratification of neuroendocrine neoplasms, with a focus on those arising in the lung and thymus, pancreas, and intestines.
Key points
- •
Neuroendocrine neoplasms arise throughout the body. They are recognized pathologically based on characteristic morphologic patterns and immunoexpression of neuroendocrine differentiation markers.
- •
The pathologic classification of neuroendocrine neoplasms has evolved over the past decades, as new understanding of the biological behavior, histologic characteristics, and genetic features of these neoplasms has emerged.
- •
Many aspects of the classification systems remain confusing or controversial. The reasons for the lack of uniformity in approach include the diversity of neuroendocrine neoplasms, the functional status of some neuroendocrine neoplasms, and the organ-specific differences.
- •
Recent efforts to standardize the classification of gastroenteropancreatic neuroendocrine neoplasms have been reasonably successful; but other organ systems, such as the lung and thymus, use different terminology and classification criteria.
- •
Genetic findings have not only helped establish relationships among different types of neuroendocrine neoplasms but they have also revealed potential therapeutic targets. Thus, the pathologic approach to neuroendocrine neoplasms is becoming more consistent and clinically relevant.
Introduction
Neuroendocrine neoplasms arise throughout the body. They are recognized pathologically based on characteristic morphologic patterns and immunoexpression of neuroendocrine differentiation markers. The pathologic classification of neuroendocrine neoplasms has evolved over the past decades, as new understanding of the biological behavior, histologic characteristics, and genetic features of these neoplasms has emerged. Nonetheless, many aspects of the classification systems remain confusing or controversial. The reasons for the lack of uniformity in approach include the diversity of neuroendocrine neoplasms. Although their shared neuroendocrine differentiation suggests a closely related family, it is now clear that several distinct types of neuroendocrine neoplasms exist. Most importantly, the well-differentiated neuroendocrine tumor (WD-NET) and poorly-differentiated neuroendocrine carcinoma (PD-NEC) families are increasingly recognized to be very different and, in all likelihood, not closely related. Other variables include the functional status of some neuroendocrine neoplasms, which can drive their clinical manifestations and treatment, relative to the nonfunctional counterparts. Finally, there are organ-specific differences.
Recent efforts to standardize the classification of gastroenteropancreatic neuroendocrine neoplasms, first proposed by the European Neuroendocrine Tumor Society (ENETS) and then adopted by the World Health Organization (WHO), have been reasonably successful; but other organ systems, such as the lung and thymus, use different terminology and classification criteria; even within the gastroenteropancreatic group there exists biological heterogeneity that is partially obscured by the standardization of classification criteria. Despite these difficulties, much progress has been made in determining the features predicting behavior. In particular, recently implemented grading schemes can effectively stratify the indolent, moderately aggressive, and highly aggressive groups of neuroendocrine neoplasms. Genetic findings have not only helped establish relationships among different types of neuroendocrine neoplasms but they have also revealed potential therapeutic targets. Thus, the pathologic approach to neuroendocrine neoplasms is becoming more consistent and clinically relevant. This review summarizes the current approach to the diagnosis, classification, grading, and therapeutic stratification of neuroendocrine neoplasms, with a focus on those arising in the lung and thymus, pancreas, and intestines. The array of rare neuroendocrine neoplasms affecting other epithelial organs and the skin (Merkel cell carcinoma) is beyond the scope of this review.
Introduction
Neuroendocrine neoplasms arise throughout the body. They are recognized pathologically based on characteristic morphologic patterns and immunoexpression of neuroendocrine differentiation markers. The pathologic classification of neuroendocrine neoplasms has evolved over the past decades, as new understanding of the biological behavior, histologic characteristics, and genetic features of these neoplasms has emerged. Nonetheless, many aspects of the classification systems remain confusing or controversial. The reasons for the lack of uniformity in approach include the diversity of neuroendocrine neoplasms. Although their shared neuroendocrine differentiation suggests a closely related family, it is now clear that several distinct types of neuroendocrine neoplasms exist. Most importantly, the well-differentiated neuroendocrine tumor (WD-NET) and poorly-differentiated neuroendocrine carcinoma (PD-NEC) families are increasingly recognized to be very different and, in all likelihood, not closely related. Other variables include the functional status of some neuroendocrine neoplasms, which can drive their clinical manifestations and treatment, relative to the nonfunctional counterparts. Finally, there are organ-specific differences.
Recent efforts to standardize the classification of gastroenteropancreatic neuroendocrine neoplasms, first proposed by the European Neuroendocrine Tumor Society (ENETS) and then adopted by the World Health Organization (WHO), have been reasonably successful; but other organ systems, such as the lung and thymus, use different terminology and classification criteria; even within the gastroenteropancreatic group there exists biological heterogeneity that is partially obscured by the standardization of classification criteria. Despite these difficulties, much progress has been made in determining the features predicting behavior. In particular, recently implemented grading schemes can effectively stratify the indolent, moderately aggressive, and highly aggressive groups of neuroendocrine neoplasms. Genetic findings have not only helped establish relationships among different types of neuroendocrine neoplasms but they have also revealed potential therapeutic targets. Thus, the pathologic approach to neuroendocrine neoplasms is becoming more consistent and clinically relevant. This review summarizes the current approach to the diagnosis, classification, grading, and therapeutic stratification of neuroendocrine neoplasms, with a focus on those arising in the lung and thymus, pancreas, and intestines. The array of rare neuroendocrine neoplasms affecting other epithelial organs and the skin (Merkel cell carcinoma) is beyond the scope of this review.
General features of neuroendocrine neoplasms
Neuroendocrine differentiation in tumors is conceptually defined as the secretion by the neoplastic cells of bioactive substances, usually bioamines or peptide hormones, into the bloodstream. Non-neoplastic neuroendocrine cells, which are dispersed within the epithelium of most organs and clustered in islets of Langerhans in the pancreas, produce similar substances; their morphologic appearance is shared by the cells of neuroendocrine neoplasms, WD-NETs in particular. The origin of neuroendocrine neoplasms from normal neuroendocrine cells has, thus, been postulated, although the concept that neoplasms arise from their mature non-neoplastic cellular counterparts is likely overly simplistic. Potentially, it is more primitive cells with stem cell features that give rise to these neoplasms, and it is the differentiation, rather than the cell of origin, of the neoplasm that allows its classification. Pathologically, neuroendocrine differentiation is defined as architectural and cytologic patterns reminiscent of non-neoplastic neuroendocrine cells (such as a nesting or trabecular growth pattern and coarsely stippled nuclear chromatin ( Fig. 1 )) and the production of characteristic neurosecretory proteins that can be detected by immunohistochemistry. A wide array of peptide hormones and bioamines can be produced as well; but for the purposes of pathologic diagnosis, it is the so-called general neuroendocrine markers that are detected. The most specific general neuroendocrine markers in wide use are chromogranin A and synaptophysin. Staining for one or both of these can be detected in essentially all WD-NETs. Other general neuroendocrine markers are available, such as CD56 (neural cell adhesion molecule) and neuron-specific enolase; but these label other types of neoplasms without known neuroendocrine differentiation and are, therefore, considered less reliable. In many cases typical examples of WD-NETs are readily recognizable as having neuroendocrine differentiation based on their routine histologic features, and immunolabeling for chromogranin A and synaptophysin is not absolutely required for their diagnosis.
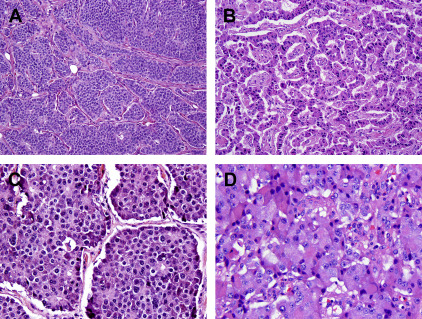
Although WD-NETs closely resemble non-neoplastic neuroendocrine cells, PD-NECs are high-grade carcinomas that exhibit neuroendocrine differentiation. These neoplasms share some histologic features with WD-NETs, but they are obviously less differentiated. Although PD-NECs typically express the same general neuroendocrine markers described earlier, the staining may be less intense and more focal. PD-NECs are usually classified as a small cell carcinoma or large cell neuroendocrine carcinoma (LCNEC), variants that are distinguished based on the cell size and nuclear morphology. Small cell carcinomas have round to fusiform cells with very little cytoplasm and hyperchromatic nuclei with a finely granular chromatin pattern and inconspicuous or absent nucleoli. The cells are often arranged in sheets, although a nested pattern can occur; there is usually single cell or geographic necrosis and a very high mitotic rate ( Fig. 2 ). The histologic features of small cell carcinoma are sufficiently distinctive that the entity can be diagnosed without the need to demonstrate neuroendocrine differentiation by immunohistochemistry, although most cases (85%) do express chromogranin or synaptophysin. LCNECs more typically demonstrate a nested architecture and are composed of larger cells with moderate cytoplasm and round or oval nuclei with more open chromatin and prominent nucleoli ( Fig. 3 ). The necrosis and high mitotic rate of small cell carcinomas is also present in LCNECs. This histologic pattern is not entity defining, however. LCNECs must demonstrate immunoexpression of at least one neuroendocrine marker to be distinguished from poorly-differentiated carcinomas of an exocrine type, such as poorly-differentiated adenocarcinoma or large cell undifferentiated carcinoma. PD-NECs are primarily distinguished from WD-NETs by having a substantially higher proliferative rate, although there are many other differences.
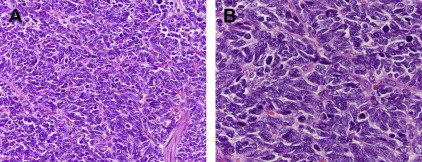
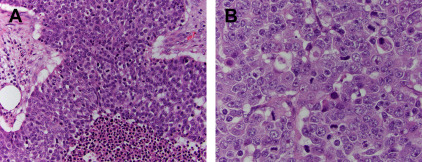
The relationship between WD-NETs and PD-NECs is becoming clearer. Although classification systems that include both entities seem to suggest that they represent the opposite ends of a spectrum of neuroendocrine neoplasms, and they do share neuroendocrine differentiation and histologic features associated with the neuroendocrine phenotype, accumulating evidence demonstrates that WD-NETs and PD-NECs are in fact two very different families of neoplasms. Several lines of evidence support this distinction. WD-NETs are generally relatively indolent and may be surgically curable if detected early, and their evolution can take years to decades when they recur; PD-NECs are highly aggressive, usually progressing rapidly even when detected at an early stage. WD-NETs and PD-NECs are etiologically different in some organs, such as the lung where small cell carcinomas and LCNECs have a close association with smoking that is lacking in carcinoid tumors (WD-NETs). Also, it is usually only WD-NETs that arise in patients with neuroendocrine neoplasia syndromes (eg, multiple endocrine neoplasia 1 [MEN1] or von Hippel Lindau syndromes). PD-NECs, at least small cell carcinomas, exhibit marked but transient sensitivity to platinum-based chemotherapy; but WD-NETs are usually resistant to platinum and other cytotoxic chemotherapies. PD-NECs often arise in association with exocrine-type precursor lesions, such as adenomas in the large intestine or ampulla of Vater, or they may be combined with adenocarcinoma or squamous cell carcinoma components, as in mixed adenocarcinoma neuroendocrine carcinoma (MANEC). WD-NETs rarely have exocrine components. Also, individual tumors containing both WD-NET and PD-NEC components are exceedingly rare. Finally, there are distinct molecular alterations in these two families of neuroendocrine neoplasms. Although some alterations are specific to the site of origin (see later discussion), WD-NETs lack alterations in genes, such as RB1 and TP53 , that are commonly found in PD-NECs.
Thus, the evidence is strong that WD-NETs and PD-NECs must be distinguished whenever possible. The distinction can be challenging in some instances, such as when only biopsy samples are available; but a pathologic diagnosis that only indicates a neuroendocrine neoplasm with specifying the differentiation is considered inadequate to direct therapy. It should also be noted that the concept of differentiation differs from that of grade, because it signifies a specific category of neuroendocrine neoplasm rather than a degree of aggressiveness. Although most classification schemes use 3 grades, current thinking is that only 2 categories of neuroendocrine tumors exist (WD-NETs and PD-NECs); there is no longer a definable group of moderately differentiated neoplasms.
The terminology for neuroendocrine neoplasms has been problematic for various reasons. Even following significant recent efforts to standardize the terminology, different terms are used in different organs to describe the same category of neoplasm. Historically, the term carcinoid tumor , coined by Oberndorfer in 1907, has been used for WD-NETs; in the pancreas, islet cell tumor was used. Currently, in the gastroenteropancreatic system, these terms have been replaced with NET to emphasize that a carcinoid tumor is a not benign neoplasm. However, this concept is not used for neuroendocrine neoplasms of the lung and thymus; also, in other rare sites (gallbladder, kidney, larynx, and so forth), the term carcinoid tumor persists. Although terminology systems vary by organ ( http://meetinglibrary.asco.org/content/115000092-156 ; Table 1 ), the distinction between the well and poorly-differentiated categories applies throughout the body.
| Differentiation | Grade | Lung | Gastrointestinal Tract and Pancreas |
|---|---|---|---|
| Well differentiated | Low grade | Carcinoid tumor | WD-NET, grade 1 |
| Intermediate grade | Atypical carcinoid tumor | WD-NET, grade 2 | |
| Poorly differentiated | High grade | Small cell carcinoma | PD-NEC, grade 3; small cell carcinoma |
| Large cell neuroendocrine carcinoma | PD-NEC, grade 3; large cell neuroendocrine carcinoma | ||
| Combined | High grade | Combined small cell carcinoma (with adenocarcinoma or squamous cell carcinoma) | Mixed adenocarcinoma neuroendocrine carcinoma (small cell carcinoma) |
| Combined large cell neuroendocrine carcinoma (with adenocarcinoma or squamous cell carcinoma) | Mixed adenocarcinoma neuroendocrine carcinoma (large cell neuroendocrine carcinoma) |
A major objective of pathologic classification is to stratify neuroendocrine neoplasms by prognosis. Many different pathologic findings have been shown to correlate with outcomes, and some are incorporated in staging systems that were developed for the first time in the most recent American Joint Committee on Cancer’s (AJCC) staging system (2009), some neuroendocrine neoplasms being staged using the same parameters as exocrine carcinomas of the same organ, others having unique NET-specific staging systems. In some organs, immunohistochemical labeling for various markers has prognostic significance, and even cytogenetic or molecular features can predict outcomes in certain circumstances. But apart from staging, the emphasis has been on grading neuroendocrine neoplasms, and grading systems based largely on the proliferative rate have been developed for thoracic and gastroenteropancreatic neuroendocrine neoplasms. Proliferative rate can be determined by counting mitotic figures (usually expressed as the number of mitoses in 10 high power microscopic fields or 2 mm 2 ) or, in the gastroenteropancreatic organs, by measuring the percentage of tumor cells immunolabeling for the proliferation marker Ki67 (the Ki67 index). The thoracic system does not use the Ki67 index, but the presence of necrosis is included. In both of these major grading schemes, neuroendocrine neoplasms are divided into 3 grades, with the low and intermediate grades (grade 1 and 2) being WD-NETs, and the high-grade (grade 3) group generally consisting of PD-NECs.
The grading parameters for neuroendocrine neoplasms of the entire gastroenteropancreatic system have been unified, such that a single system proposed by ENETS and endorsed by the WHO is now widely used for these primaries ( http://meetinglibrary.asco.org/content/115000092-156 ; Table 2 ). In the lung and thymus, a different WHO-accepted system has been in place for many years ( http://meetinglibrary.asco.org/content/115000092-156 ; Table 3 ). For organs outside of these sites, no formal systems exist; but individual proposals have been based on the thoracic or gastroenteropancreatic systems. The major differences between the thoracic and gastroenteropancreatic systems are as follows:
- 1.
The use of necrosis to separate low- from intermediate-grade WD-NETs in the thorax
- 2.
The requirement for Ki67 staining in the gastroenteropancreatic system
- 3.
The different mitotic rate cut point that defines high grade (10 mitoses per 10 high power fields in the thorax; 20 mitoses per 10 high power fields in gastroenteropancreatic organs)
| Tumor Grade | Definition |
|---|---|
| Low grade (grade 1) | <2 mitoses per 10 HPF and Ki67 index <3% |
| Intermediate grade (grade 2) | 2–20 mitoses per 10 HPF or Ki67 index 3%–20% |
| High grade (grade 3) | >20 mitoses per 10 HPF or Ki67 index >20% |
| Neoplasm | Morphology | Mitoses | Necrosis | Immunohistochemistry |
|---|---|---|---|---|
| Typical carcinoid tumor | Polygonal cells arranged in nested or trabecular patterns | <2 per 10 HPF | Absent | Chromogranin, synaptophysin, CD56 (supportive but not required) |
| Atypical carcinoid tumor | Polygonal cells arranged in nested or trabecular patterns | 2–10 per 10 HPF | Present, usually punctate | Chromogranin, synaptophysin, CD56 (supportive but not required) |
| Large cell neuroendocrine carcinoma | Large cells, moderate cytoplasm, round nuclei, frequent nucleoli | >10 per HPF | Present, usually extensive | Chromogranin, synaptophysin, CD56 (at least one required) |
| Small cell carcinoma | Small cells, scant cytoplasm, fusiform nuclei, no nuclei | >10 per HPF | Present, usually extensive | Chromogranin, synaptophysin, CD56 (supportive, but not required) |
The ability of these systems to stratify the outcome of neuroendocrine neoplasms has been convincingly demonstrated for multiple anatomic sites. Interestingly, the outcome difference between low grade and intermediate grade is striking, even though the proliferative rate that distinguishes these groups is very modest (0–1 mitoses per 10 high power fields vs 2 or more). To some extent, the grade correlates with stage; but these prognostic parameters are independently predictive of outcome. Even among patients with stage IV disease, the grade stratifies the length of survival. Thus, the importance of accurate grading is now well accepted for neuroendocrine neoplasms, even though there may not presently be major management differences between low- and intermediate-grade NETs. It should be emphasized that the initial proposed grading parameters were chosen based on the experience and intuition of the individuals who proposed the systems, rather than on rigorous review of outcome data. The specific cut points of mitotic rate and Ki67 index chosen to separate the grades are continually reevaluated and may require adjustment as data accumulate. Also, the attempts to unify the grading parameters for many anatomic sites are laudable; but it is clear that distinctive features specific to each site of origin exist, and organ-specific grading systems may prove necessary for optimal stratification.
Grading neuroendocrine neoplasms requires precise determination of mitotic rate and Ki67 index. These assessments have proven more challenging than they might seem. The mitotic rate is expressed as a number per unit area (10 high power fields), but in fact the diameter of a high power microscopic field varies among microscopes. In an effort to render this more reproducible, some investigators have used an actual area (2 mm 2 ), which corresponds to the size of 10 fields on one of the most standard microscopes in current use. But this means that pathologists using other microscopes would need to adjust their counting based on measuring the field area, a correction that is not often performed.
Another obvious issue is that within the family of WD-NETs, there is considerable variation in cell size and stromal content, both of which affect the number of cells within a given area. There has been no proposal to correct for these variables; but in theory a tumor composed of large cells with more abundant stroma would have fewer mitotic figures identified than a densely cellular tumor with minimal cytoplasm, even though the actual proportion of cells undergoing division were the same. These vagaries settle out when examining the outcome of a large patient cohort; but for the individual case, they can lead to underestimation of grade.
The use of the Ki67 index corrects for these issues because the index is expressed as a percentage of positive-labeling nuclei, rather than a number per unit area. In order to calculate the Ki67 index, it was originally recommended to count the regions with the greatest proportion of positive nuclei (hot spots). Heterogeneity of labeling is common, and data comparing the average labeling rate with that of the hot spots confirm that the more proliferative regions more accurately predict the outcome. Nonetheless, the exact size of a hot spot is uncertain (one 200 × microscopic field has been proposed). Furthermore, the identification of hot spots requires that a large sample of the tumor be stained for Ki67.
When biopsy samples are evaluated, this is not possible. The theoretic risk that biopsies may, therefore, underestimate the grade of WD-NETs has been proven, using virtual randomly oriented biopsies of resected hepatic metastases. A single core biopsy from a G2 WD-NET has only a 35% chance of accurately identifying the higher proliferative rate; 3 core biopsies identify the G2 focus in 48%, and it would take 31 core biopsies to identify 90% of G2 cases. Conversely, an apparent G1 WD-NET graded on a core biopsy has only a 59% chance of truly being low grade. The phenomenon of grade heterogeneity exists within the primary WD-NET, between the primary and metastases, and also between different foci of metastatic disease. A higher-grade component can also emerge during the course of disease progression. Targeting a metastasis that shows radiographic evidence of growth may be a more reliable way to ensure that the highest-grade focus is detected.
Another consideration, for gastroenteropancreatic NETs, is that the mitotic rate and Ki67 index may point to different grades. In one study of pancreatic WD-NETs, 107 of 297 tumors (36%) had discordance between these two proliferation indices. Usually it is the Ki67 index that suggests the higher grade, and the original grading system proposed by ENETS recommended relying on whichever measure defined the higher grade. This suggestion has also been supported by data showing that WD-NETs with discordant mitotic rate and Ki67 index (G1 by mitoses, G2 by Ki67) behave more like cases whereby both indices point to the higher grade.
A final consideration regarding Ki67 is the means to assess the index itself. The ENETS and WHO recommend counting 2000 cells (or at least 500 cells) to accurately determine the Ki67 index. It is very challenging to undertake this count while reviewing the actual glass slide, and many pathologists have been simply estimating the percentage by casual observation (eyeballed estimate). Although eyeballing is simple and fast, the degree of interobserver and intraobserver variability using eyeballing is unacceptably high.
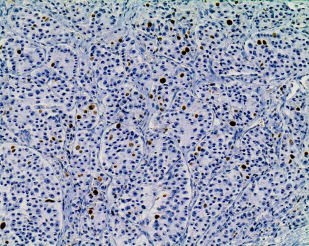
Digital image analysis can be used to determine a highly accurate percentage, provided that the instrument is calibrated to recognize truly positive cells and non-neoplastic cells, whether positive or negative, are excluded from analysis. Digital image analysis has been shown to correlate almost perfectly with manual counting, but it is not in routine use and requires considerable time to perform properly. Therefore, many experts suggest manual counting based on a printed photomicrograph of the hot-spot regions ( Fig. 4 ). All of these issues have called into question the entire concept of measuring proliferation to grade NETs, but the practice is now well established; the resulting grades have strong prognostic implications, even with all of the challenges.
Related posts:
 Neuroendocrine Tumors—Current and Future Clinical Advances
Neuroendocrine Tumors—Current and Future Clinical Advances
 Surgical Treatment of Small Bowel Neuroendocrine Tumors
Role of Somatostatin Analogues in the Treatment of Neuroendocrine Tumors
Surgical Treatment of Small Bowel Neuroendocrine Tumors
Role of Somatostatin Analogues in the Treatment of Neuroendocrine Tumors
 Neuroendocrine Tumors—Current and Future Clinical Advances
Systemic Therapies for Advanced Pancreatic Neuroendocrine Tumors
Neuroendocrine Tumors—Current and Future Clinical Advances
Systemic Therapies for Advanced Pancreatic Neuroendocrine Tumors
 Surgical Management of Pancreatic Neuroendocrine Tumors
Surgical Management of Pancreatic Neuroendocrine Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


