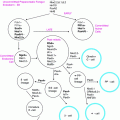
Fig. 18.1
Mutations in a number of genes can lead to a variety of syndromes of dysgenesis involving the Müllerian or Wolffian ducts, gonads, kidneys, and adrenal glands as a result of a deficiency or excess of the proteins shown. DAX1 denotes the gene duplicated in congenital adrenal hypoplasia on the X chromosome; Emx2, the empty spiracles homeobox gene; GATA-4, the gene encoding a protein that binds to a GATA DNA sequence; HOXA, homeobox protein; Lim1, a homeobox gene important for limb development; Lhx9, a lim homeobox family member; Pax2, a paired box homeotic gene; SF-1, the gene for steroidogenic factor 1; SRY, the sex-determining region of the Y chromosome; SOX9, SRY homeobox 9; WNT-4, a protein that induces development of the Müllerian mesenchyma; and WT1, Wilms’ tumor suppressor gene 1
Male and female primordial reproductive ducts coexist for a short period in all mammalian embryos. Müllerian ducts are the anlagen of the uterus, Fallopian tubes, and upper third of the vagina, and develop autonomously in the female in the absence of the testis. Wolffian ducts are the anlagen of the epididymis, vas deferens, and seminal vesicles, and require testosterone to develop. In the mouse, the Wolffian duct requires the action of PAX2 and PAX8, paired genes first discovered in Drosophila. Müllerian duct formation requires the activity of a series of secreted factors whose expression is directed by the WNT family of proteins [10, 11]. The Müllerian duct first forms by invagination of the coelomic epithelium which tubularizes under the influence of Wnt4, then elongates, as the epithelial cells migrate in close approximation to the Wolffian duct [12]. The Wolffian duct provides no cells, but its presence is essential for full elongation of the Müllerian duct to the distal urogenital sinus [13].
Primordial germ cells, the progenitors of the oocytes and spermatocytes, migrate to the urogenital ridge from outside the embryo [14]. They travel from the embryonic ectoderm, along the primitive streak, to the base of the allantois, the wall of the hindgut, and finally exit into the urogenital ridge, where they take up residence in the as yet undifferentiated gonad. Ectopic germ cells outside the urogenital ridge either fail to develop or form tumors [15, 16]. The migratory germ cell phenotype is controlled by integrins [17] and cKit signaling [18] and it is characterized by extensive mitosis [19]. The germ cells proliferate under the influence of Blimp-1 [20], BMP 2,4, and 8b, and Fragilis. Fragilis induces Stella that in turn induces the pluripotency genes Oct/4, Sox2, and Nanog. Germ cell expansion can be detected by expression of alkaline phosphatase and Stella [21, 22] as early as 3 weeks gestation in the human [23]. In addition to proliferation, the germ cells undergo imprint erasure imposed by DNA methylation and complex histone modification to reset epigenetic memory [24–26]. After entering the mesonephros, female germ cells enter meiosis in a characteristic anterior to posterior wave that is under the influence of retinoic acid mediated by the recently discovered gene, stimulated by Retinoic Acid 8 (Stra8). Since the testes degrade retinoic acid, Stra8 is not expressed, meiosis is not initiated, and male germ cells remain arrested in the G0 phase of the cell cycle [27, 28].
The early, undifferentiated gonads exist as a primitive blastema of mesenchymal cells covered by coelomic epithelium. Testicular morphogenesis is initiated by a short burst of expression of the small transcription factors SRY, a master switch gene present on the short arm of the Y chromosome, and the SRY-related autosomal gene SOX 9 from chromosome 17q. The fact that full gonadal differentiation beyond the streak gonad stage requires the Y chromosome or the second X chromosome became evident in 1959 when the 45, X Turner phenotype was first recognized [29]. Subsequent cytogenetic studies localized the male differentiation region to the short arm of the Y chromosome [30, 31]. Later, a single-copy gene, the sex-determining region of the Y chromosome (SRY), was identified that encodes a DNA-binding protein, which is expressed in the gonadal ridge immediately before testis differentiation [32, 33]. When the SRY gene was transfected into XX female mouse embryos, a substantial proportion of these transgenic animals developed testes and assumed anatomic and functional male phenotypes [32, 34].
In addition to the Y chromosome, autosomal and X chromosome factors contribute to sex differentiation. For example, mutations on the long arm of chromosome 17q, found in cases of campomelic dysplasia associated with sex reversal [35–37], caused a defect in the SRY-related gene product, SOX 9, which contributes to sex differentiation. In addition, analysis of 46, XY sex-reversed females with intact SRY led to the discovery of the dosage-sensitive sex (DSS) reversal locus on the short arm of the X chromosome, duplication of which is required for female differentiation [38]. This region, which contains the gene DAX1, was then assumed to have a negative influence on testis differentiation because a double dose of the X chromosome is associated with dysgenesis of the testis. However, knockout of DAX1 resulted in normal female gonadal development [39] which undermines the hypothesis that DAX1 is responsible for ovarian differentiation. The only abnormality seen as a result of null mutation in DAX1 occurs, counter intuitively, in the male, which displays abnormalities in spermatogenesis and spermatic cord formation [40]. Defects in the distal end of the short arm of chromosome 9p [41] and the distal end of the long arm of chromosome 10q [42] are also associated with sex reversal.
The genes responsible for ovarian development remain relatively obscure. Previously, the conventional wisdom was that the ovaries developed passively as a result of the absence of testicular determining genes. It is now clear that Wnt–4 is associated with ovarian development, as homozygous inactivation of Wnt–4 in females leads to masculinized gonads and an absence of the Müllerian ducts [11]. Foxl2, a forkhead transcription factor, is another gene that represses male development, allowing an XY gonad to develop as an ovary [43, 44]. Prenatal ovarian development also appears to be independent of steroid hormone action [45]. Taken together, it is now clear that ovarian differentiation is not simply a default pathway in the absence of testis differentiation [46].
The fetal testis develops seminiferous tubules with Sertoli cells surrounding germ cells and interstitial Leydig cells. The fetal testis produces two products required for further male differentiation, Müllerian Inhibiting Substance (MIS), which inhibits differentiation of the Müllerian duct [47], and testosterone, which stimulates Wolffian structures [47]. The external genital primordia develop autonomously into clitoris, labia minora, and labia majora. Complete differentiation of the external genitalia to phallus and scrotum requires reduction of testosterone to dihydrotestosterone by 5α-reductase [48]. The interaction of dyhydrotestoterone and androgen receptors causes lengthening of the phallus into a penis, fusing of the urogenital folds to form the penile urethra, and fusion of the labioscrotal swellings in the midline to form the scrotum. Autonomous female development can occur in the absence of ovaries.
The existence of a Müllerian inhibitor was proposed by Jost [47], who showed that testicular implants in female rabbit embryos stimulated the Wolffian duct but also caused regression of Müllerian ducts that could not be recapitulated by implants of testosterone alone. This regression is characterized morphologically by programmed cell death. Müllerian Inhibiting Substance (MIS) was purified [49, 50], and then used to clone the MIS gene [51, 52]. The bioactive C-terminal domain of MIS is homologous to a group of evolutionary conserved proteins, referred to as the transforming growth factor-ß (TGF-ß) family [52]. The MIS ligand binds to a heterologous receptor composed of at least two serine–threonine kinase transmembrane units, the type II receptor [53], which phosphorylates or activates the type I receptor [54], which in turn signals downstream via SMAD1/5/8 to initiate a series of molecular events that results in regression of the Müllerian ducts. MIS has been developed as an antiproliferative agent targeting tumors of Müllerian duct origin such as ovarian [23, 55], endometrial [56], cervical [57], cancers, as well as breast [58–60], and prostate [58] cancers. Abnormalities of the MIS gene itself or its receptors can result in the retained Müllerian duct syndrome, in which otherwise normal males, usually with undescended testes, have persistent Müllerian structures that have not undergone normal regression [61, 62].
Pathophysiology of Disorders of Sexual Differentiation (DSD)
There are three major categories of developmental aberrations that are responsible for the most common forms of Disorders of Sex Development (DSD) in newborns (Table 18.1). In the first category of DSD, the external genitalia of genetic females are masculinized by an excessive androgenic steroid production. In the second category of DSD, genetic males have deficient androgen production or action. The third category of DSD is notable for gonadal differentiation that is absent, incomplete, or asymmetrical [4]. It is important for physicians involved in the care of patients with DSD to understand the underlying pathophysiology so that they can make a timely and accurate diagnosis and be able to advise optimal treatment strategies.
Table 18.1
Disorders of sex development diagnosis and treatment
Disease | Diagnostic features | Physical examination phenotype | Gender assignment | Medical therapy | Surgical therapy |
|---|---|---|---|---|---|
46, XX DSD (overandrogenized female) Congenital adrenal hyperplasia (adrenogenital syndrome) | Karyotype 46, XX electrolytes: K high, Na low Androgen high 17-hydroxyprogesterone high MIS 0 Sequence CYP21 Chromosomal FISH | Symmetrical gonads Clitoral hypertrophy Sinogram: UG sinus defect Enlarged labioscrotum | F | Hydrocortisone or cortisone acetate + Florinef Embryo selection from the blastocyst Steroid replacement in utero from 6 wk gestation | Perioperative stress steroids Clitoral reduction Vaginal exteriorization Labioscrotal reduction |
46, XY DSD (Underandrogenized male) Testosterone deficiency | Karyotype 46, XY Testosterone low Sequence enzyme genes: 17-KS OH, P450scc, 3β-HSD, CYP17 FISH | Symmetrical, undescended, small testis Severe hypospadias No müllerian structures | If M | Presurgical testosterone stimulation Testosterone at adolescence | Hypospadias repair Prepenile scrotal repair Orchiopexy |
Predominant female phenotype if adolescence | If F | Estrogen/progesterone at | Gonadectomy | ||
Androgen receptor deficiency Testicular feminization (complete androgen insensitivity) | Karyotype 46, XY Testosterone high MIS high Sequence androgen receptor gene FISH | Female phenotype No müllerian structures Normal testes Narrow male pelvic structures Sparce axillary and pubic hair | F | Estrogen/progesterone at adolescence | Infancy: gonadectomy, Adolescence: vaginal replacement |
Reifenstein’s syndrome (incomplete androgen insensitivity) | Karyotype 46, XY Testosterone normal MIS slightly elevated | As for testosterone if deficiency | If F | Estrogen/progesterone at adolescence | Infancy: gonadectomy, clitoral reduction, labioscrotal reduction Adolescence: vaginal replacement |
Sequence androgen receptor gene FISH CGH | If M | As for testosterone deficiency (above) | AS for testosterone deficiency (above) | ||
5α-Reductase deficiency | Karyotype 46,XY Testosterone high DHT low MIS high Sequence 5α-reductase type 2 gene FISH | Severe hypospadias Symmetrical, undescended, normal testes Prepenile scrotum | M | Presurgical testosterone stimulation DHT replacement | Hypospadias repair Prepenile scrotal repair Prostatic and utricle opening |
46, XY (complete gonadal dysgenesis) | Karyotype 46, XY Sequence candidate genes: SRY, SOX 9, DAX-1 Testosterone absent MIS absent FISH CGH | Phenotypic female Vagina present No gandas Symmetrical | F | Estrogen/progesterone at adolescence | Gonadectomy |
45, X/46, XY (Ovotesticular DSD; MGD (mixed gonadial dysgenesis) | Karyotype 45, X/46, XY or 46, XY Testosterone low MIS low Sequence DAX-1 FISH CGH | Asymmetry of gonads: streak ovary and dysgenetc testis UG sinus Clitoral hypertrophy | If F | Estrogen/progesterone at adolescence | Clitoral recession Vaginal exteriorization Labioscrotal reduction Gonadectomy |
If M | Presurgical testosterone stimulation Testosterone at adolescence | ||||
46 XY (90%) Ovotesticular DSD True hermaphrodite | Karyotype 46, XX Testosterone normal or low MIS normal or low CGH | Asymmetrical testis, ovary, or ovotestes UG sinus defect Clitoral hypertrophy | If F | Estrogen/progesterone at adolescence | Clitoral recession Vaginal exteriorization Labioscrotal reduction Preserve normal ovary or polar ovarian tissue |
If M | Presurgical testosterone stimulation Testosterone at adolescence | Staged hypospadias repair Prepenile scrotal repair Removal of müllerian structures Preserve vas and normal testis or central testicular tissue Orchiopexy? Prosthesis? |
46, XX DSD, Overandrogenization of the Genetic Female
In 46, XX DSD excessive androgens lead to virilization (or masculinization) of genetic females. Patients with 46, XX DSD were previously described as female pseudohermaphrodites or labeled with the diagnosis of congenital adrenal hyperplasia (CAH) or adrenogenital syndrome [63, 64]. The most common cause of excessive androgens is a defect in one of the P450 enzymes that in the adrenal gland convert progesterones to glucocorticoids and mineralocorticoids (Fig. 18.2). These enzyme abnormalities may occur in males and females but they cause ambiguous genitalia only in females.
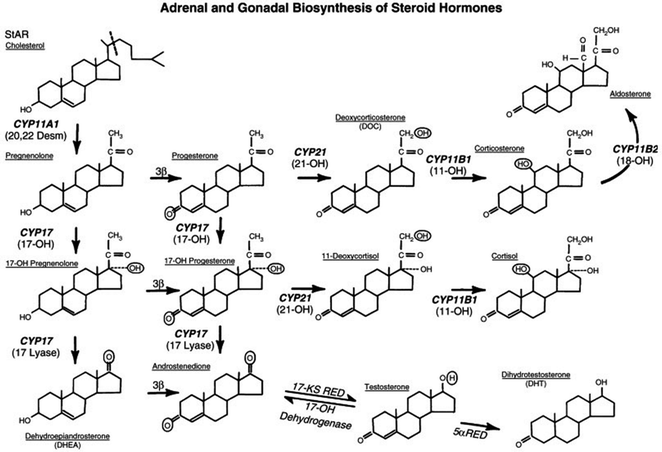
Fig. 18.2
Pathways of steroid hormone biosynthesis from cholesterol. New enzyme nomenclature is in bold lettering; old nomenclature is in parentheses. Enzymatic defects of CYP21, CYP11, or 3ß-hydroxysteroid dehydrogenase can lead to congenital adrenal hyperplasia. Deficiencies in testosterone production leading to a form of male pseudohermaphroditism can result from defects in CYP11A1, CYP17, 3ß-hydroxysteroid dehydrogenase, and 17-ketosteroid reductase
In the human fetus adrenal differentiation occurs at 11 weeks gestation, after differentiation of the gonads and reproductive tract. Therefore, these enzyme defects lead to an excess androgen accumulation after 11 weeks gestation and affects the external genitalia that develop after that time—the genital tubercle and the urogenital folds. The genital tubercle normally forms the clitoris, but when exposed to excess androgens, develops as a penile structure. The urogenital folds, rather than developing as labia, acquire a bifid unfused or only partially fused scrotal appearance.
Mutations in the P450c21, or 21-hydroxylase gene, now known as CYP21 (Table 18.2), cause 90% of cases of CAH. The remainder of CAH cases are distributed among deficiencies of P450c11, 3ß-hydroxysteroid dehydrogenase, CYP17, or Steroid Acute Regulatory Protein (StAR), depending on the ethnic origin of the patient [4, 65, 66]. Rarely, 46, XX DSD results from exposure to exogenous androgens.
Table 18.2
Steroid biosynthetic enzyme nomenclature
Old | New-protein | New-gene |
|---|---|---|
Testis | ||
20,22-desmolase | P450scc (side chain cleavage) | CYP11A |
17-hydroxylase | P450c17 | CYP17 |
17-lyase | P450c17 | CYP17 |
3β-hydroxysteroid | Same | Same |
dehydrogenase (3ß-HSD) | ||
17-ketosteroid reductase | Same | Same |
Adrenal | ||
21-hydroxylase | P450c21 | CYP21 |
11-hydroxylase | P450c11 | CYP11B1 |
18-hydroxylase | P450c11 | CYP11B2 |
All the enzymatic defects of CAH result in deficient cortisol biosynthesis that stimulates the pituitary gland to increase corticotropin production. The increase in corticotropin stimulates the adrenal gland and leads to adrenal hyperplasia, elevated production of hormonal precursors proximal to the enzymatic defect, and preferential overproduction of androgenic steroids (Fig. 18.2). Elevations in 17-hydroxyprogesterone can be detected rapidly from a spot serum sample [67] which is mandatory in some states. Polymerase chain reaction (PCR), sequencing of known genes can be used to make this diagnosis in utero [4].
46, XY DSD, Undervirilization of the Genetic Male
Insufficient virilization (masculinization) of a 46, XY genetic male can occur because of insufficient androgen production, an androgen receptor deficiency, or an inability to convert testosterone to dihydrotestosterone. The three distinct subtypes of 46, XY DSD are reviewed below. 46, XY DSD was formerly known as male pseudohermaphroditism [68].
Testosterone Deficiency
Deficiency of androgen production can be caused by genetic defects of the enzymes [CYP11A1 (20,22 desmolase), CYP17 (17-hydroxylase or lyase), 3β-hydroxysteroid dehydrogenase (3ß-HSD), and 17β-hydroxysteroid dehydrogenase (17ß-HSD), or 17-ketosteroid reductase], which together and sequentially are responsible for the metabolism of cholesterol to testosterone (Fig. 18.2) [65]. When stimulated by chorionic gonadotropin patients with defects of these enzyme produce no (or only a minimal amount of) testosterone and because of testosterone deficiency, the penis may be small and hypospadiac [69]. In contrast, MIS levels are normal or high for their age and result in an absence of Müllerian structures. The testes may be undescended, small, or both.
Androgen Insensitivity
Androgen insensitivity is the most common cause of 46, XY DSD. Most abnormalities are caused by point mutations in the androgen receptor gene [70, 71]. However, not all patients with androgen insensitivity have a molecular defect in the androgen receptor gene. In these patients it is possible that the noncoding promoter, the 3′ untranslated region, or other transcription factors or their cofactors might be the cause of 46, XY DSD. There are two known isoforms of the androgen receptor. Type 2 is the predominant receptor in the external genitalia while the type 1 receptor is found in a multitude of other tissues [10].
The phenotype of androgen insensitivity can be mild or severe. The complete androgen insensitivity syndrome (CAIS), previously known as testicular feminization, results in a female phenotype even though the serum testosterone is high. MIS may be normal or, in some cases, considerably elevated, therefore Müllerian structures usually regress normally. The gonads are symmetrical and may be intra-abdominal or descended.
5α-Reductase Deficiency
Mutations of 5α-reductase type 2 genes lead to testosterone not being converted to the active hormone dihydrotestosterone in peripheral, particularly genital, target tissues [48, 72]. Normal dihydrotestosterone action in the genital area results in elongation and ventral closing of the penile raphe, which encloses the urethra and displaces the urethral orifice from the perineum to the tip of the penis [68] and in addition causes the labioscrotal folds to fuse to create scrotal sacs. The autosomal recessive 5α-reductase type 2 deficiency causes severe hypospadias associated with undescended testis, prepenile scrotum, and an enlarged prostatic utricle. The enzyme has binding sites for both testosterone and an NADPH cofactor. Point mutations in the NADPH cofactor have been associated with the defective phenotype of 46, XY DSD. The increase in activity of the 5α-reductase type 1 isoform at puberty produces a paradoxical virilization that may result in a change of sexual identity.
Abnormalities of Gonadal Development and Differentiation
Abnormalities of the sex chromosomes usually manifest as failed, incomplete, or asymmetrical gonadal differentiation. Patients with these disorders have either bilateral streak gonads, as in 46, XY pure gonadal dysgenesis DSD, or asymmetrical gonadal development, as in mixed gonadal dysgenesis (MGD) DSD or ovotesticular DSD (formerly termed true hermaphroditism).
46, XY Pure Gonadal Dysgenesis DSD
Patients with 46, XY pure gonadal dysgenesis DSD may have a defective Y chromosome [73]. Other causes of 46, XY pure gonadal dysgenesis DSD include mutations associated with camptomelic dysplasia, a severe disorder occurring in patients with a translocation in the distal arm of chromosome 9p near the SRY-related SOX 9 gene [35, 37], and mutations in WT1 associated with Frasier’s syndrome [41]. The streak gonads in 46, XY pure gonadal dysgenesis DSD fail to develop bilaterally. They produce little or no testosterone so gonadotropin levels are compensatorily high. The streak gonads also produce no MIS so Müllerian duct structures are preserved resulting in a female phenotype.
Mixed Gonadal Dysgenesis (MGD) DSD
MGD DSD (also known as asymmetric gonadal dysgenesis) with a 45,X/46,XY karyotype is by far the most common of the chromosomal abnormalities causing DSD [4, 74]. The gonads are asymmetrical, most often with a small dysgenic testis on one side and a streak gonad on the other [75]. Most patients with this defect have retained Müllerian ducts. The small testis can produce enough testosterone to cause masculinization and hypertrophy of the clitoris. The vagina fails to migrate to the perineum and enters the urethra as a urogenital sinus (UGS) defect more distal to the bladder neck than seen in severe cases of CAH.
It is important to note that 40% of patients with MGD can have a 46, XY karyotype, and some have bilateral testes or streak ovaries. MGD is poorly understood at the molecular level [76]. The absence of the second X chromosome is in some way related to the early ovarian dysgenesis. The mosaicism of mixed gonadal dysgenesis results from the presence of at least two gonadal (chimeric) germ cell lines. The degree of testicular differentiation is determined by the percentage of cells expressing the XY genotype, which may also influence the degree of asymmetry. Loss of the Y chromosome can occur because of nondisjunction, the failure of paired chromosomes to migrate to opposite poles during cell division [1, 29, 77, 78].
Ovotesticular DSD
True ovotesticular DSD (previously known as true hermaphrodism) is rare, except among the Bantu in Southern Africa [79, 80]. More than 90% of these patients have a 46, XX karyotype. Asymmetry characterizes many of these patients, who have simultaneous ovarian and testicular differentiation without the dysgenesis characteristic of MGD. The etiology of ovotesticular DSD and the reason for the gonadal asymmetry remains an enigma. The testicular and ovarian tissue can be separated (i.e., an ovary on one side and a testis on the other), or can occur as ovotestis on both sides, or be combined only in one gonad as an ovotestis with a normal ovary or a testis on the other. When ovarian tissue and testicular tissue coexist in the same gonad, the testicular tissue is always central, and the ovarian tissue is polar [81]. Although early testicular differentiation occurs, spermatogenesis is not advanced [79], which may reflect the absence of other necessary Y-directed functions. The Müllerian structures are regressed on the side of the testicular tissue but retained on the side of the ovarian tissue and in the midline as well, with the vagina entering the distal urethra creating a UGS defect.
Diagnosis
A baby born with DSD must have a thoughtful yet expeditious evaluation to determine whether gender assignment can be made. An important goal is to minimize the immediate and long-term emotional trauma to the family and child and this is best accomplished by early referral to an experienced team of endocrinologists, geneticists, psychologists, and pediatric surgical specialists.
Although many syndromes can affect later sexual development, only four DSDs present at birth: (1) 46, XX DSD, formerly known as female pseudohermaphroditism, congenital adrenal hyperplasia (CAH) or adrenogenital syndrome [63, 64], which causes overandrogenization of a genetic female, (2) 46, XY DSD, previously termed male pseudohermaphroditism, which causes undervirilization of a genetic male, (3) MGD DSD mixed gonadal dysgenesis 45, X/46, XY, or (4) ovotesticular DSD, formerly known as true hermaphroditism. A child with pure gonadal dysgenesis, although having a 46, XY karyotype, is phenotypically female.
The diagnostic evaluation of patients with ambiguous genitalia is outlined in Table 18.1. Two screening criteria can be used to diagnose the infant as having one of the four disorders, symmetry and the presence of a Y chromosome (Table 18.3). The first diagnostic criterion is the physical finding of gonadal symmetry or asymmetry, and the second is sex chromosome determination. Gonadal symmetry is defined by the position each gonad relative to the external inguinal ring; i.e., gonads are symmetric when both are either above or below the external inguinal ring and gonads are asymmetric when one gonad is above the external ring and the other gonad is below the external inguinal ring. Gonads are symmetrical in 46, XX DSD and 46, XY DSD when a systemic biochemical defect influences both gonads equally. Gonadal asymmetry occurs in chromosomal abnormalities such as MGD (mixed gonadal dysgenesis) or ovotesticular DSD (true hermaphroditism), in which a predominant testis descends below the external ring and a predominant ovary remains above the external ring.
Table 18.3
Rapid diagnostic algorithm
Y Chromosome absent or abnormal | Y Chromosome present | ||
|---|---|---|---|
Gonadal symmetry | Gonadal asymmetry | Gonadal symmetry | Gonadal asymmetry |
46, XX DSD[congenital adrenal hyperplasia] | Ovotesticular DSD | 46, XY DSD | 46, XX/46, XY MGD [Mixed gonadal dysgenesis] |
A detailed history and physical examination can be coupled and genetic testing to define the mutations or deletions known to cause defects in the four major categories and to define the DSD more fully. Subsequently, ultrasonography, magnetic resonance imaging, contrast radiography, and later panendoscopy, coupled with accurate laboratory analysis for disorders of enzymes affecting testosterone synthesis, should provide a definitive diagnosis and permit appropriate gender assignment with a high degree of accuracy.
The size of the penis and its location with respect to the scrotum should be carefully noted. Although the size of the phallus varies considerably, some guidelines are helpful [82, 69]. The average length of the penis is 3.5 ± 0.4 cm at term, 3.0 ± 0.4 cm at 35 weeks’ gestation, and 2.5 ± 0.4 cm at 30 weeks’ gestation. At term, a diameter of 1 cm is average. A penis smaller than 1.5 ± 0.7 cm in a full-term infants raises the possibility of female gender assignment, except in cases of 17-ketosteroid reductase or 5α-reductase deficiencies, when large body habitus or sex reversal at puberty, respectively, would favor male sex assignment. An early rectal examination permits detection of a uterus still enlarged under the influence of placental gonadotropin. The presence and severity of hypospadias should be noted. Palpation of the gonads can differentiate the firm testis from the softer ovotestis and differentiate a testis with an attached epididymis from an ovary in the inguinal position [4].
46, XX DSD Overandrogenization of the Genetic Female
If the patient is the proband for the family, the diagnosis of 46, XX DSD can be delayed but if a previous sibling has already manifested the disorder then genetic testing could permit prenatal diagnosis.
The condition presents in a wide clinical spectrum. The ovaries, uterus, and Fallopian tubes are normal. The vagina, however, is foreshortened because of failure to migrate to the perineum. Instead the vagina usually merges with the urethra to form a urogenital sinus (UGS) distal to the bladder neck. However, a rare but significant subset of patients may have a verumontanum, with the urethrovaginal confluence quite close to the bladder neck [83]. These severely masculinized patients can also have an enlarged, placentally stimulated prostate, palpable at the level of the verumontanum.
The external genitalia are characterized by variable clitoral enlargement, ranging from trivial to severe. In severe cases patients have an almost normal male phallus. The labia can be masculinized to form either enlarged labioscrotal folds or, in the most severe cases, complete scrotal fusion. The disorder is systemic so the gonads are symmetric and because the ovaries are normal, the gonads never descend into the labioscrotal folds or fused scrotum [4, 84]. The karyotype is always 46, XX in females. MIS is undetectable and serum 17-hydroxyprogesterone is elevated. Androgen levels are also elevated. Concomitant production of melanocyte-stimulating hormone can darken the genitalia and breast areolae.
An infant with bilateral nonpalpable testes should have a rapid analysis for 17 hydroxyprogesterone (a screening test done in some states on all newborns) and a karyotype determination to identify the presence or absence of the Y chromosome. These tests should identify patients with 46, XX DSD who has CYP21 disorders that cause potential life-threatening hormonal deficiencies.
PCR with appropriate probes demonstrates point mutations in the P450c21 gene [85]. Electrolytes are often normal at birth, but when maternal steroids wane after 5–7 days, serum sodium levels may fall and serum potassium levels may become markedly elevated which if unattended, can lead to cardiac arrest. Hence, this disorder must be considered a medical emergency until treatment is commenced.
46, XY DSD, Undervirilization of the Genetic Male
Testosterone Deficiency
By definition, 46, XY DSD patients have deficient androgenization of the external genitalia with a 46, XY karyotype. If the disorder is caused by an enzymatic defect in the production of testosterone, patients have low basal serum testosterone, and stimulation by chorionic gonadotropin produces little or no increase in testosterone. Because the genes coding for the enzymes in the pathway have been cloned, PCR or direct sequencing can be performed to detect specific deficiencies. The testes may be small and are often bilaterally and symmetrically undescended. The penis is small, and hypospadias is usually severe. Since MIS is normal there are no detectable Müllerian structures. In some cases, the phenotype is completely female [69, 86]. Gender assignment is controversial. The size of the phallus plays a role, as it may be better to raise infants with a small, nonreconstructable phallus as females, whereas it is invariably preferable to raise those with a reasonably sized phallus as males, particularly if they respond to exogenous testosterone. Special consideration should be given to those rare children with 17-ketosteroid reductase deficiency, which may be better raised as males because of later large body habitus.
Androgen Insensitivity
Patients with severely dysfunctional androgen receptors have Complete Androgen Insensitivity Syndrome (CAIS), previously known as testicular feminization [87]. The phenotype is female and these children have been raised as females. No Müllerian structures are present because the normal testes produce high levels of biologically active MIS that regress the Müllerian ducts. The testes are usually in the inguinal region, and their firmness and attached epididymis distinguish them from ovaries. The karyotype is 46, XY. The androgen receptor is deficient and the normal testes to produces high levels of testosterone and MIS. PCR or sequencing of the androgen receptor gene can often pinpoint the molecular defect and provide the definitive diagnosis.
Partial Androgen Insensitivity Syndrome (PAIS) results in only partially masculinized 46, XY patients, with a wide variation in phenotype. As with testosterone-deficiency syndrome, the penis is small and hypospadiac. The testes may be small and are often undescended but symmetrical, and Müllerian structures are not present. The testosterone levels are normal, and MIS levels are elevated. Most abnormalities are caused by point mutations in the androgen receptor gene [70, 71]. Again, the size of the phallus can influence gender assignment.
5α-Reductase Deficiency
46, XY DSD can also result from a deficiency of 5α-reductase, which is responsible for the conversion of testosterone to dihydrotestosterone which acts on the genital area resulting in elongation and ventral closing of the penile raphe, with normal displacement of the urethral orifice from the perineum to the tip of the glans penis [68]. This process is abnormal in these patients, resulting in severe penoscrotal hypospadias. Normal and symmetrical testes can be either undescended or fully descended. The labioscrotal folds are also closed posteriorly to create partial or bifid scrotal sacs. The prostatic utricle is often quite enlarged. Again, the karyotype is 46, XY. Serum levels of testosterone are high, but levels of dihydrotestosterone are low. The MIS levels are normal and Müllerian structures are not present. PCR and full DNA sequencing can be used to genotype and to detect mutations in the 5α-reductase type 2 gene.
At puberty 5-α-reductase type I present in skin and other organs markedly increase resulting in a dramatic phenotypic conversion and it is now considered more appropriate to raise these children as males. It should be noted, however, that in this group, gender assignment involves the greatest dilemma in societies committed to two sexes, but not in societies that more readily accept assignment to a third sex [48, 88, 89].
Abnormalities of Gonadal Development and Differentiation
46, XY Pure Gonadal Dysgenesis
Patients with pure gonadal dysgenesis are born as phenotypic females and there are no questions of ambiguity. Amniocentesis with a 46, XY karyotype that does not produce the expected phenotype on fetal ultrasonography allows earlier diagnosis than was previously possible. Dorsal pedal edema and some Turner characteristics may be the only obvious somatic manifestations of the defect in otherwise normal females. Müllerian structures are present, but gonads are not palpable due to failure of gonadal differentiation. Dysgenesis of the gonad results in an absence of testosterone and MIS.
Mixed Gonadal Dysgenesis (MGD)
Patients with mixed gonadal dysgenesis are characterized by asymmetry, with a streak gonad on one side and a dysgenetic testis on the other. Because the testes are dysgenetic, testosterone and MIS levels may be low. They have retained Müllerian structures because of lack of MIS [74, 75, 90]. The clitoris is usually hypertrophied. The most common karyotype is 45, XO/46, XY, but 40% of patients have the 46, XY karyotype. There is a propensity for neoplastic transformation of the abnormal gonads to gonadoblastoma or seminoma (or both) [78, 91, 92], that may occur even in newborns. These tumors can cause gonadal torsion and apparent loss of a gonad that, in rare cases, leads to a unilateral dysgenetic testis or streak ovary. Because gonadoblastomas may occur at any age, gonadectomy and female reconstruction and rearing are the options usually chosen. There is a subgroup of 45, XO/46, XY patients that are diagnosed prenatally with normal external genitalia and raised as males indicating a spectrum of the disorder.
Related posts:
 Assessing and Selecting Patients for Bariatric Surgery
Diabetes in the Pediatric Surgical Patient
Assessing and Selecting Patients for Bariatric Surgery
Diabetes in the Pediatric Surgical Patient
 Parathyroid Gland Embryology, Anatomy and Physiology
Parathyroid Gland Embryology, Anatomy and Physiology
 Surgical Considerations of the Pituitary
Surgical Considerations of the Pituitary
 Ovarian Embryology, Anatomy, and Physiology Including Normal Menstrual Physiology
Ovarian Embryology, Anatomy, and Physiology Including Normal Menstrual Physiology
 Pancreas Embryology, Anatomy, and Physiology
Pancreas Embryology, Anatomy, and Physiology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


