xenografts, cancer stem cells were found to be enriched in the surviving tumor tissue.10,11 These surviving cancer stem cells were found to possess fewer reactive oxygen species (ROS) (mediators of radiation-induced damage) and activated DNA damage response/repair pathways in response to the radiotherapy. Knowledge of the biologic nature and response of stem cells has led to the hope of targeting these resistance mechanisms therapeutically.
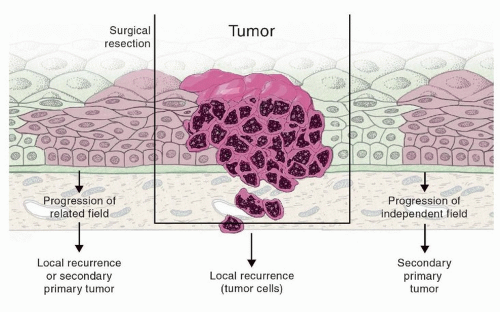 Figure 2.1. Field cancerization. Field cancerization is defined as the presence of one or more mucosal areas consisting of epithelial cells that have cancer-associated genetic or epigenetic alterations. A preneoplastic field (shown in light pink) is monoclonal in origin and does not show invasive growth or metastatic behavior, which are the hallmarks of an invasive carcinoma (dark pink). Field cancerization has been supported by molecular data and provides a theoretical explanation for multiple primaries. (Adapted from Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11(1):9-22.) |
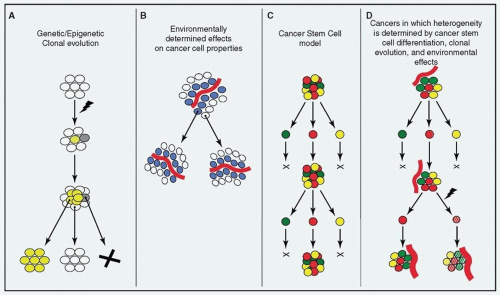 Figure 2.2. Tumor heterogeneity. Heterogeneity can arise within tumors through: (A) the stochastic process of clonal evolution, (B) extrinsic environmental differences within tumors, and (C) the presence of cancer stem cells that variably differentiate. These processes are not mutually exclusive, but rather synergistic in producing a heterogeneous population of cells (D). (From Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21(3):283-296, with permission.) |
may be principally responsible13,14 for creating a heterogeneous population of cells, but that clonal evolution and the effects of the tumor’s microenvironment act in a synergistic manner (i.e., the cancer stem cells themselves may undergo clonal evolution and be affected by local influences in the tumor environment). Higher levels of intracancer heterogeneity have also been correlated to tumor progression, poorer survival, and adverse outcomes in patients with HNSCC. Recently, levels of tumor heterogeneity, when factored with HPV status, were found to be useful in predicting clinical outcome15 in HNSCC.
Table 2.1 Distinct Clinical Features of HPV(+) and HPV(-) HNSCC | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
tobacco.28,29 In addition to the direct trauma to mucosal surfaces induced by these agents, tobacco products are composed of dozens of known carcinogenic compounds, including polycyclic aromatic hydrocarbons (PAHs), oxidizing substances, and free radicals. Following metabolic activation by endogenous enzymes (often cytochrome p450s), these carcinogens form covalent DNA adducts and/or induce epigenetic changes. These DNA adducts must be repaired by designated DNA repair machinery or else risk causing errors in replication (resulting in mutations).
DNA replication or repair, exposure to mutagens/carcinogens, or infection by microorganisms and viruses. The initiation and progression of cancer involves a stepwise accumulation of these genetic insults (or “hits”). These “hits” are usually alterations in tumor suppressor genes or oncogenes. They can accumulate in many forms, including mutations, copy number variations (CNVs), epigenetic changes, and others. We will discuss the alterations that contribute to development or progression of HNSCC (Fig. 2.3). While genes such as p53 and p16 are altered in the vast majority of HNSCCs, most affected genes in
HNSCC occur in fewer than 30% of the cancers. Despite the enormous number of potential alterations, they tend to cluster in a limited number of biologic pathways, which helps to organize and understand the pathogenesis, and will be discussed afterward.
Table 2.2 Familial Syndromes and HNSCC | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
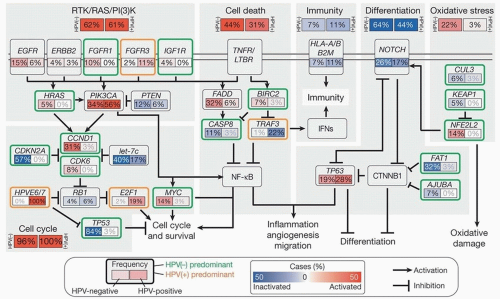 Figure 2.3. Pathways affected in HNSCC. Signaling pathways frequently altered in HNSCC, based on recent TCGA analysis. The frequency (%) of genetic alterations for HPV(-) and HPV(+) tumors is shown separately within subpanels and highlighted. (From The Cancer Genome Atlas N. Comprehensive genomic characterization of squamous cell carcinoma of the head and neck. Nature. 2015;517(7536):576-582, with permission.) |
Table 2.3 Common Gene Defects in HNSCC | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
By altering the nonsequence structure of DNA and the histones that package it, epigenetic modifications can make genes more or less accessible to activators of transcription and therefore modulate their expression. Methylation is the most frequently studied epigenetic change, partly because there are well-established methods to examine it. Promoter methylation, as well as global hypermethylation, has been shown to facilitate tumorigenesis by the silencing of tumor suppressors. In HNSCC, promoter methylation of genes such as CDKN2A (encoding p16INK4a and p14ARF), DAPK, RASSF1A, RARB2, APC, and MGMT is often an early event during neoplastic progression.47,48,49 It has been postulated that epigenetic changes are complementary to the genetic changes, for example, silencing a wild-type tumor suppressor allele when the other is inactivated by mutation.
by DNA repair machinery, bringing the final error rate to one per ten million bases. A wide variety of systems known as “caretakers” detect and repair errors in the genome or, if unsuccessful, freeze the cell cycle and activate cell death.61,62 Some mechanisms directly inactivate or intercept mutagens even before they damage DNA. The “clonal evolution theory” suggests that many alterations are needed for cells to gradually transform into cancer. Although the chances of this happening in a normal cell are infinitesimally small, damage to caretakers through genetic or epigenetic changes can lead to instability of the genome, increasing the error rate and allowing propagation of defects to subsequent generations of cells. Thus, by allowing the series of alterations required for neoplastic change, genomic instability is considered an enabling characteristic common to nearly all cancers.63 The quintessential tumor suppressor p53 plays a central role in guarding the genome by activating DNA repair, cell cycle arrest, or apoptosis in response to genetic damage or cellular stresses.64
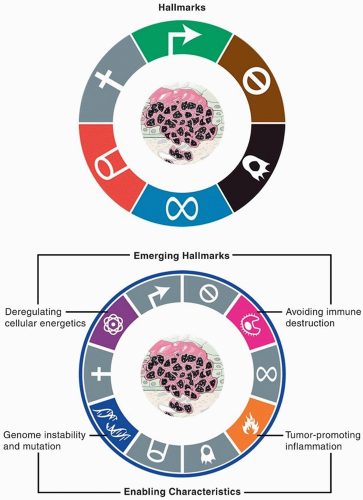 Figure 2.4. Hallmarks of cancer. Hanahan and Weinberg initially described, then expanded, attributes needed for cancer development. (Adapted from Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.) |
slough away. The normal maturation of epithelial cells involves orderly changes as they progress from the basal epithelial layer to become mature keratinocytes (Fig. 2.6). These changes are mediated by transcriptional changes driving increased or decreased expression of specific genes involved in epithelial differentiation. Disruption of this normal differentiation is thought to occur in HNSCC and other epithelial cancers, in which cell populations can attain properties and appearance of mesenchymal (connective tissue) cells. The resulting cells often lose their epithelial architecture, become less dependent on cell-cell contact, and possess increased motility, invasion, angiogenesis, and other mesenchymal-like properties. This phenomenon is referred to as epithelial-mesenchymal transition (EMT). The process of EMT is mediated by four major transcription factors: Snail, Slug, Twist, and Zeb 1/2. EMT is a normal process that is used during embryogenesis and wound healing but is hijacked during carcinogenesis.
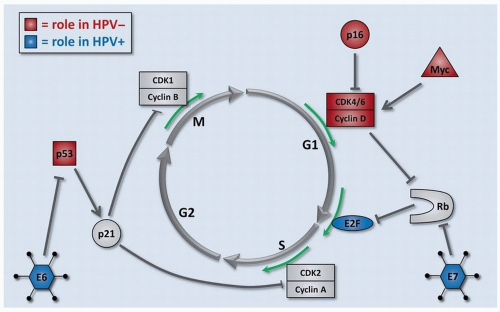 Figure 2.5. Cell cycle control. Progression through the cell cycle is tightly controlled in noncancer cells. In HNSCC, the regulators of cell cycle progression are ubiquitously altered by many different mechanisms. The most common defects in cell cycle regulators are shown for HPV- (red) and HPV+ (blue) HNSCCs. |
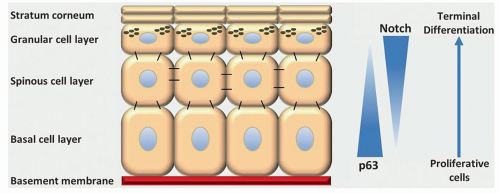 Figure 2.6. Maturation of keratinocytes. Epithelial cell proliferation is limited to the basal layer, and cells progressively differentiate as they migrate superficially before being sloughed from the surface. NOTCH and p63 play key roles in this differentiation process and are frequently disrupted during development of HNSCC. |
(Fig. 2.7).72,73 These changes are needed for the destructive invasive properties of HNSCC as well as for processes associated with lymphatic and hematogenous metastases.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


