4 Pathogenesis and Pathology of Diabetic Neuropathy
Histopathology
 Diabetic Peripheral Neuropathy
Diabetic Peripheral Neuropathy
A.P. Mizisin and H.C. Powell
The frequent occurrence of neurologic complications of diabetes has long been recognized and no doubt contributed to the erroneous belief of nineteenth-century physicians that diabetes mellitus was a disease of the nervous system. While disturbances in the central nervous system related to insulin deficiency are recognized, the major neurologic complication is the peripheral neuropathy occurring in both insulin-dependent and insulin-independent forms of diabetes mellitus. Although conventional medical treatment prolongs life span and attenuates neurologic complications of diabetes, hyperglycemic control is not sufficient to prevent the development of neuropathy. The peripheral nerve disorders related to diabetes mellitus are clinically heterogeneous and often subdivided into symmetric polyneuropathies and focal or multifocal neuropathies.
The pathology of diabetic neuropathy and its interpretation have been a continuing source of controversy. Points of contention have ranged from whether peripheral nerve injury is primary or secondary to neuronal degeneration to whether demyelination or axonal loss is the primary or main lesion. The pathogenesis of diabetic neuropathy has also been contentious and variously described as having a metabolic or ischemic etiology. Despite disagreement about the primary role of a particular lesion or the etiology of diabetic neuropathy, it is clear that diabetes mellitus has the potential to induce pathologic changes in most cellular and noncellular components of the peripheral nerve. This chapter will consider first the histopathologic changes induced by hyperglycemia in the peripheral nerve and then the relationship of this pathology to the type of diabetic neuropathy.
Hyperglycemia-Induced Histopathology
Myelinated Nerve Fibers
Loss of myelinated nerve fibers has been repeatedly documented. While fiber loss is most prominent distally, it may also be apparent in spinal roots, particularly in dorsal roots. Some have suggested that proximal multifocal fiber loss in the sciatic nerve summates to produce diffuse distal lesions in the peroneal, tibial, and sural nerves [1]. Although marginal fiber loss is difficult to assess qualitatively, moderate to gross loss has been extensively illustrated (Fig. 4.1a), often with considerable variation between adjacent fascicles. A diabetes-induced decrease in the density and occupancy of myelinated fibers represents quantitative evidence of loss affecting both large and small fibers [2–4].
Changes noted prior to the axoplasmic dissolution that constitutes axonal degeneration include accumulation of glycogen and dystrophic accumulation of vesicular and cytoskeletal elements [5,6]. Demyelination secondary to axonal degeneration has been observed [7]. Characteristic of axonal degeneration of the Wallerian type, osmiophilic lipid droplets can be observed within otherwise vacant neurilemmal tubes in teased fiber preparations (Fig. 4.2a). The Schwann cell basal laminae that form neurilemmal tubes are frequently circular, as if failing to collapse, and assume the corrugated profile seen in typical Wallerian degeneration [8]. In earlier stages of diabetic neuropathy, axonal regeneration has been reported to be robust and greater than that in control subjects [2]. Regenerative clusters appear in plastic sections as a group of myelinated sprouts within a residual, circular basal lamina [5,8].
In human diabetic neuropathy, the existence of axonal atrophy or the diminution of axonal caliber without myelin or axonal degeneration is disputed. Axonal atrophy was suggested by an early report of teased fibers with long internodes and inappropriately small diameters [9]. However, despite qualitative descriptions [5] and quantitative evidence employing multiple parameters [4,10,11], axonal atrophy has not been observed in other studies [12,13], including one involving a large sample size and claiming an improved morphometric method for detecting this change [14].
Segmental demyelination has long been described as a pathologic change occurring in diabetic neuropathy [2,9,15,17]. It is recognized in teased fibers as an internode lacking myelin or with an inappropriately thin myelin sheath compared to the myelin surrounding the adjacent internodes (Fig. 4.2b). In plastic section, demyelination was described as splitting of myelin sheaths with accumulation of granular and vesicular debris [5]. Early reports of segmental demyelination without prominent axonal degeneration no doubt contributed to the view that demyelination is the primary lesion of diabetes-induced nerve injury. However, some investigators [9] noted that certain clinical features of diabetic neuropathy were best explained as resulting from a combination of segmental demyelination and axonal degeneration. Indeed, both primary segmental demyelination and demyelination secondary to axonal degeneration have been documented in the same nerve biopsy [7].
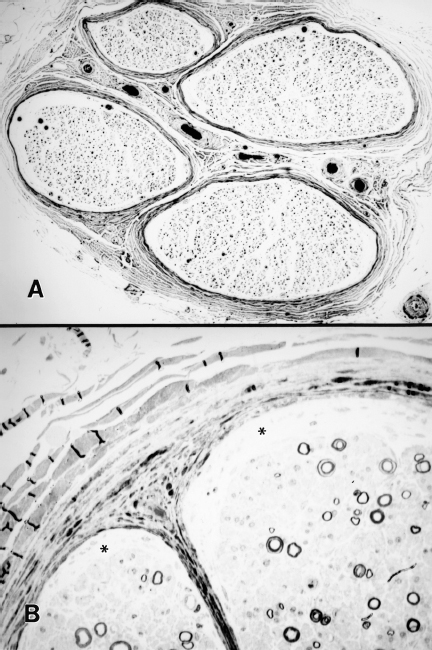
Fig. 4.1 Myelinated fiber loss in chronic human diabetic neuropathy. A A sural nerve biopsy shows fascicles with severe fiber loss. Several fascicles also show increased subperineurial structureless space, consistent with endoneural edema. B Higher magnification view of a plastic section from a sural nerve showing subperineurial edema (asterisks) and myelinated fiber loss
Schwann cell changes that appear to precede the dissolution of the myelin sheath have been observed in human diabetic neuropathy by several investigators [5,6,18]. Nonspecific, reactive changes include: accumulation of lipid droplets, paracrystalline inclusions (Pi granules of Reich) and glycogen granules: increased numbers of plasmalemmal vesicles; and cytoplasmic expansion and capping (Fig. 4.3a). Enlarged mitochondria with effaced cristae and disintegration of abaxonal and adaxonal cytoplasm and organelles have been described as degenerative Schwann cell changes (Fig. 4.3b). Thickening and reduplication of the Schwann cell basal lamina of myelinated fibers have also been illustrated [6].
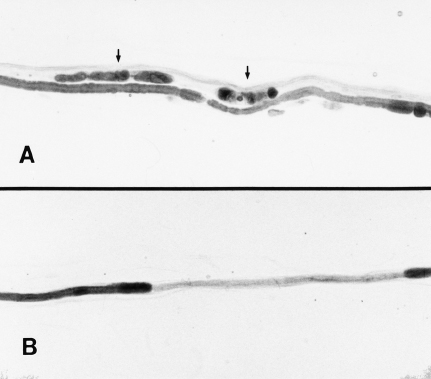
Fig. 4.2 Pathological abnormalities of teased nerve fibers from sural nerve biopsies in human diabetic neuropathy. A Wallerian degeneration, characterized by nerve fiber breakdown and consequent formation of myelin ovoids (arrows), is seen above an intact myelinated fiber. B A teased fiber from another biopsy shows an internode with severe myelin loss, consistent with either segmental demyelination or early remyelination. (Micrographs kindly provided by Nigel A. Calcutt, PhD)
Remyelination following segmental demyelination has been observed in diabetic neuropathy and is recognized in teased fiber preparations [9,16] and plastic section [12,18,19] by axons with inappropriately thin myelin sheaths. In some but not all nerve biopsies, proliferative Schwann cell changes are evident as clusters of Schwann cells in a concentric arrangement (Fig. 4.4) [5,9,20]. These concentric arrangements resemble small “onion bulbs,” a nonspecific hypertrophic change consisting of supernumerary Schwann cell processes surrounding individual axons. “Onion bulbs” are thought to result from recurrent segmental demyelination and remyelination [20].
Paranodal abnormalities described in diabetic neuropathy include demyelination, paranodal swelling and axo-glial dysjunction. Several investigators [4,9,10] have emphasized the occurrence of restricted paranodal demyelination (Fig. 4.5a), which may be resolved with selective remyelination by surviving Schwann cells or with the formation of an intercalated internode as noted in teased fibers [2]. Paranodal swelling has been suggested to precede paranodal demyelination and is thought to be associated with axo-glial dysjunction, the loss of the gap-junction-like connections of terminal Schwann cell loops to the axolemma on either side of the node of Ranvier [4]. The existence of paranodal swelling and axo-glial dysjunction is a contentious issue. Although repeatedly documented by some in experimental and human diabetic neuropathy [4,10], others [21] have not detected these abnormalities.
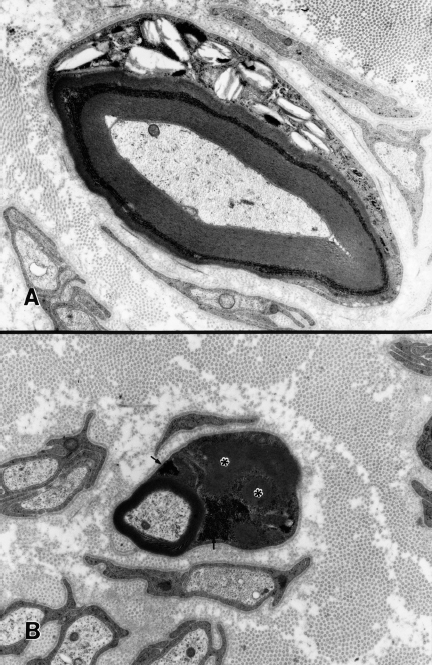
Fig. 4.3 Reactive and degenerative Schwann cell changes in poorly controlled human diabetic neuropathy. A Lysosomal inclusions (Pi granules of Reich), a nonspecific reactive change characteristic of chronic neuropathies with extensive myelinated fiber loss, are evident in an intemodal band of Schwann cell cytoplasm in this myelinated fiber. B A small myelinated fiber with degenerative changes shows conspicuous Schwann cell cytoplasmic enlargement with glycogen accumulation (arrows) and darkened profiles of giant mitochondria with effaced cristae (asterisks)
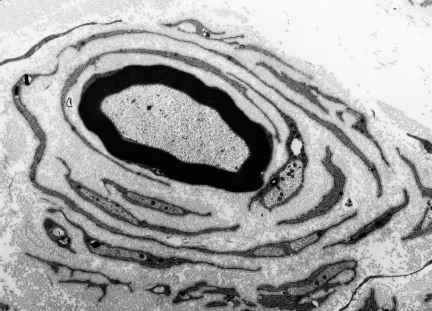
Fig. 4.4 Proliferative Schwann cell changes in chronic human diabetic neuropathy. Concentric arrays of supernumerary Schwann cells form an “onion bulb” around a myelinated fiber
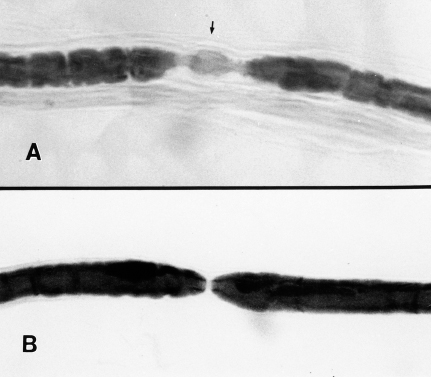
Fig. 4.5 Paranodal demyelination of a teased nerve fiber in human diabetic neuropathy. A The paranodal region (arrow) of this teased fiber is incompletely ensheathed by myelin, resulting in an exaggeration of the length of the node of Ranvier. B A normal-appearing node of Ranvier in a teased fiber is shown for comparison. (Micrographs kindly provided by Nigel A. Calcutt. PhD)
Unmyelinated Nerve Fibers
Early electron microscopic studies of diabetic neuropathy noted a distinct loss of unmyelinated fibers [6]. Characteristic degenerative changes of these fibers include shrinkage of axons, accumulation of enlarged vesicular elements, and deterioration of tubular and filamentous elements of the cytoskeleton. Edematous Schwann cell cytoplasm has also been observed, as well as hyperplasia of surrounding basal lamina [6]. Complete degeneration results in empty or denervated Schwann cell subunits surrounded by a basal lamina. It is thought that eventually the Schwann cells degenerate, leaving the basal lamina that persists before disappearing. In the sural nerve of a patient dying with diabetes mellitus, unmyelinated fiber density was only a third of that observed in control patients [3]. Although unmyelinated fiber density is a quantitative reflection of fiber loss, empty Schwann cell subunits are considered by some to be a better indicator of such loss [22].
Vasa Nervorum
The blood supply of peripheral nerve trunks, the vasa nervorum, consists of intrinsic endoneurial vessels and extrinsic vessels of the epineurium and perineurium. In diabetes mellitus, histopathologic changes have been described in all components of this vasculature. In the endoneurium, vessels with thickened walls and reduced luminal caliber were documented in an early report [23]. Subsequent qualitative and quantitative work has demonstrated endothelial cell hypertrophy and hyperplasia with a reduction in luminal size [11,24–29]. Fenestrated endothelial cells, a feature normally present only in epineurial vessels, have been observed in endoneurial vessels [26], as has endothelial cell dysjunction or the loss of junctional contacts between cells [29]. Desquamation of endothelial cells [30] and degeneration of pericytes have also been described [31]. Reduplication of the basal lamina of endoneurial microvessels, although a feature of other chronic neuropathies, appears to be more pronounced in diabetic neuropathy (Fig. 4.6a). Luminal occlusion resulting from endothelial hyperplasia or fibrin plugs has been documented [30,32,33] but not confirmed in subsequent studies [24,28,29].
With respect to the extrinsic circulation, epineurial capillary abnormalities include endothelial cell hyperplasia and thickening of the basal lamina [25]. The intima of epineurial arterioles is increased in diabetic neuropathy [34]. However, in spite of these changes, endoneurial microvessels show significantly more pathology than epineurial microvessels with respect to basal lamina thickening, endothelial cell hypertrophy, and luminal narrowing [25]. Similar findings are reported for the transperineurial circulation, with hypertrophy and hyperplasia of endothelial cells and reduced luminal area [35]. Diabetic patients exhibit a greater degree of abnormal innervation of the epineurial and transperineurial circulation in that there appears to be a reduction in the vessels with perivascular axons and an increase in vessels with denervated Schwann cell units [36]. In the media of denervated arterioles, structural changes, such as an increase in glycogen, edematous smooth muscle cells, accumulation of cellular debris, and collagenous scarring, have been reported.
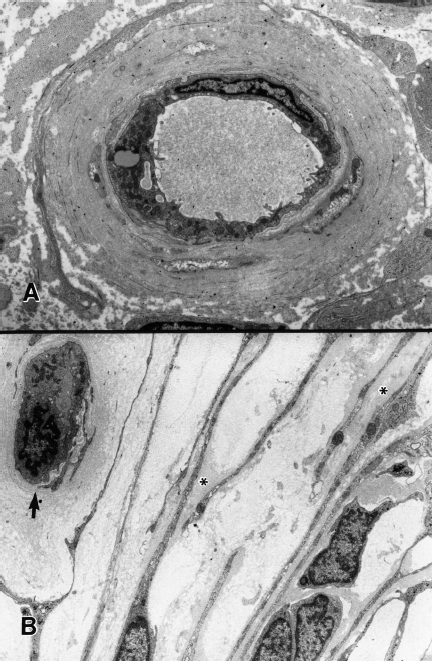
Fig. 4.6 Vascular and perineurial abnormalities in chronic human diabetic neuropathy. A Markedly thickened and reduplicated basal lamina is evident surrounding an endoneurial microvessel. B The perineurial sheath is shown with focal basal laminar thickening (asterisks). A subperineurial capillary (arrow) shows a reduplicated basal lamina
Connective Tissue
Where substantial loss of myelinated fibers was apparent in diabetic neuropathy, early workers noted fibrosis and probable increase in endoneurial collagen and, in one instance, an accumulation of fibrillar material in an enlarged endoneurial interstitium [9]. Recent work points to extensive deposition of endoneurial collagen in nerves from diabetic patients, predominantly involving collagen types I and III [37]. Type VI collagen is increased in the endoneurium surrounding groups of Schwann cells, with types IV, V, and VI increased around endoneurial microvessels. The diameter of endoneurial collagen fibrils is increased in diabetic nerves. The hyperplasia and reduplication of basal laminae surrounding Schwann cells and micro-vessels have been noted above.
An increase in endoneurial area in diabetic neuropathy has been observed in plastic section [2,18,24,38] and considered by some to represent endoneurial edema (Fig. 4.1b). In several studies using noninvasive magnetic resonance spectroscopy [39,40], hydration was increased in nerves from both asymptomatic and symptomatic diabetic patients but not in those receiving treatment with aldose reductase inhibitors. Edematous nerves appear to be an inconstant feature of diabetic neuropathy.
In diabetic patients, there are several abnormalities in the perineurium, the lamellar cellular ensheathment of individual fascicles of peripheral nerves. Thickening of the basal lamina surrounding cells of each layer of the perineurium has been documented (Fig. 4.6b) [8,41–43]. Reduplication as seen in basal laminae of Schwann cells and endoneurial microvessels is not present [43]. Calcification of the extracellular matrix of the perineurium has also been documented in diabetic neuropathy and is thought to result from deposition on matrix vesicles or lipid droplets derived from perineurial cells [44].
Relationship of Histopathology to Type of Diabetic Neuropathy
Because most cellular and noncellular components of peripheral nerves are affected in diabetes mellitus, it is difficult to ascribe a corresponding set of pathologic changes to any clinical presentation. Nevertheless, the topic has received sufficient attention in the literature to warrant consideration.
Symmetric Polyneuropathy
Among the various clinical presentations of diabetic neuropathy, distal symmetrical polyneuropathy with a “glove and stocking” distribution is the most typical. While causation remains uncertain, hyperglycemia underlies other putative mechanisms and there are some differences in patterns of structural injury in treated versus untreated patients. In treated diabetic patients with chronic neuropathy, fibers undergoing axonal degeneration predominate [45]. In contrast, in untreated diabetics with symptomatic neuropathy, both segmental demyelination and axonal degeneration are evident. Segmental demyelination appears to precede axonal degeneration and is in some instances accompanied by proliferative changes of Schwann cells including “onion bulbs” [12,18,46].
Although the most severe neuropathies are associated with profound loss of myelinated and unmyelinated axons, disturbances of lesser severity do not lend themselves to ready morphologic distinction. It appears that even in mild neuropathy, there is significant loss of myelinated fibers [19]. Although axonal regeneration may be more vigorous in milder cases of diabetic sensory neuropathy [12], it is diminished in proportion to the amount of myelinated nerve fiber loss [47]. In addition to proliferative changes of Schwann cells, marked thickening of vessel walls including thickening and reduplication of basal lamina has been found in asymptomatic patients with minimal or no signs of neuropathy [46].
Studies of painful diabetic neuropathy have investigated possible morphologic correlates of pain in patients with differing presentations of pain-related symptoms [11]. Axonal degeneration and reductions in fiber density were present in both patients with chronic neuropathy and those with diabetic pain of recent onset. In another study of patients with active acute painful neuropathy and patients with recently remitted pain [48], the occurrence of nerve fiber degeneration and regeneration was not sufficient to account fully for diabetic neuropathic pain. However, certain stages of the pathologic process of Wallerian degeneration may be linked to painful symptoms [48]. Another large clinicopathologic study also failed to establish a correlation between axonal degeneration or regeneration and painful neuropathy [12].
In severe diabetic neuropathy, autonomic disturbances are prominent but the disease is often painless. In such cases, nerve fiber loss may be profound [12]. Recurrent foot ulcers are most likely to occur in this group [11]. In the most severe neuropathies, sometimes presenting in untreated patients, demyelination and remyelination are prominent. An increase in capillary wall thickness was most pronounced in patients in whom neuropathy was painless, the degree of thickening in patients with painful neuropathy being less exaggerated [11]. With respect to capillary mural thickening, similar findings have been reported in asymptomatic patients [46].
Asymmetric Neuropathy
Focal neuropathic disorders in patients with diabetes mellitus are not symmetric in distribution and may involve cranial nerves or spinal roots. With respect to cranial nerves, the most frequently cited examples describe lesions involving the oculomotor nerve [49–51], although the trochlear and abducens nerves may be affected. Neuropathies affecting these cranial nerves are characterized by relatively sudden onset, focal distribution, and limited course [51]. Early published reports linked these neuropathies to ischemic events, and evidence of centrofascicular degeneration observed in postmortem studies reinforces this view. Nerve fiber atrophy and microfasciculation have also been documented in oculomotor nerves [51].
Proximal diabetic neuropathy is a severe form of asymmetric neuropathy that has been subject to confusing terminology and incomplete pathologic information. The clinical presentation typically involves one leg or thigh with eventual spreading to the buttock and opposite lower limb. In contrast to symmetrical polyneuropathy, this disorder appears to have an inflammatory basis [52–54]. Centrofascicular axonal loss in association with vasculitis [53] and evidence of epineurial vasculitis [54] support an ischemic causation. Occlusive vascular disease has also been detected in proximal diabetic neuropathy [53]. The presence of both axonal degeneration and demyelination attests to the role of ischemia in producing these changes in association with the inflammatory process [54].
 Diabetic Autonomic Neuropathy
Diabetic Autonomic Neuropathy
R.E. Schmidt
Neuropathology of Clinical Diabetic Autonomic Neuropathy
The neuropathology of diabetic autonomic neuropathy has, until recently, been largely unstudied despite its substantial clinical importance and wealth of pathophysiologic characterization of autonomic function in humans.
Sympathetic Ganglia
Degenerative changes culminating in the significant loss of sympathetic neurons have been claimed and disputed in classical nonquantitative studies [55–57]. Quantitative, although nonstereologic, analysis of neuronal density (expressed as number/mm2) in the diabetic paravertebral superior cervical (SCG) and prevertebral superior mesenteric ganglia (SMG) in one large series [58] showed only a small (14%), although statistically significant, decrease in neuronal density in diabetic subjects and did not identify significant numbers of actively degenerating neurons. The size and plexiform anatomy of human prevertebral ganglia and the existence of preferentially targeted subpopulations of sympathetic neurons may even complicate nonbiased stereologic analysis. Chromatolytic neurons or nodules of Nageotte (i.e., collections of satellite cell nuclei at sites of neuronal dropout) were not more common in diabetic human ganglia than in agematched nondiabetics [59]. One often-quoted study by Duchen and colleagues [55] involved the detailed histopathologic characterization of the sympathetic ganglia of five patients with symptomatic diabetic autonomic neuropathy and reported a variety of apparently pathologic findings including neuronal necrosis, an inflammatory infiltrate, neuronal gigantism, dilated perikaryal endoplasmic reticulum, vacuolated neurons, and neuroaxonal dystrophy (NAD). Unfortunately, no controls were included in the study. In a large controlled study of NAD (Fig. 4.7), the distinctive and marked enlargement of distal preterminal axons and synapses, also represented the most striking histologic alteration in diabetic sympathetic ganglia. Dystrophic swellings consisted either of disorganized neurofilamentous aggregates (Fig. 4.7) or collections of mitochondria, dense bodies, lucent proteinaceous material, and tubulovesicular elements [59]. Quantitative studies demonstrated a progressive increase in the frequency of dystrophic axons as a function of age, diabetes, and gender (males more affected than females). Diabetic patients developed lesions (immunohistochemically and ultrastructurally identical to those in aged subjects) earlier and in greater numbers than age-matched control subjects, suggesting possible shared pathogenetic mechanisms in aging and diabetes. Perikarya of diabetic principal sympathetic neurons, although compressed and distorted by presynaptic NAD, were otherwise unremarkable.
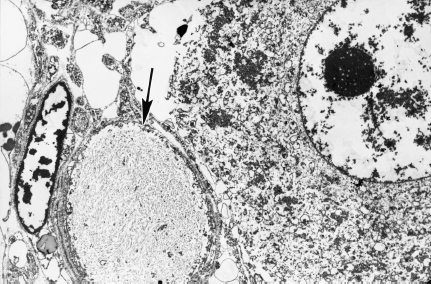
Fig. 4.7 A swollen dystrophic axon (arrow) filled with neurofilaments distorts the contours of an adjacent principal sympathetic neuron in diabetic human sympathetic SMG (magnification 3000×)
Not all sympathetic ganglia are equally affected in human diabetics. The frequency of NAD in prevertebral SMG and celiac ganglia was more than 10-fold that of the paravertebral SCG. We have reexamined (R.E. Schmidt, unpublished data) multiple prevertebral and paravertebral chain ganglia of one of Duchen’s original patients [55] who had symptomatic diabetic autonomic neuropathy with prominent alimentary dysfunction. Although the prevertebral celiac ganglia and paravertebral SCG were extensively and minimally involved, respectively, in that case the paravertebral lumbar sympathetic chain ganglia showeda frequency of NAD intermediate between those of the SCG and celiac ganglia. Prominent NAD in the celiac ganglia of the relatively young diabetics with symptomatic alimentary autonomic neuropathy [55] suggests possible pathophysiologic significance.
Lymphocytic infiltrates in postmortem sympathetic diabetic ganglia [55] have been interpreted as evidence of an autoimmune pathogenesis [60]; however, similar infiltrates were present in nearly half of all examined SCG and SMG in a large series [59] and their presence failed to correlate statistically with age, gender, or diabetes. Although the presence of antibodies against sympathetic ganglia and vagus nerve has also been reported to correlate [60] with autonomic dysfunction in diabetics, other studies have failed to show such a relationship [61].
Studies of prevertebral sympathetic ganglia in man and experimental animals have demonstrated the complexity and importance of function of prevertebral ganglion neurons in the integration of visceral reflexes [62]. Nerve terminals in the SMG reflect the contribution of neurons originating in the spinal cord intermediolateral nucleus, dorsal root ganglia, parasympathetic nervous system, other sympathetic ganglia or intraganglionic projections from neighboring principal sympathetic neurons, and from myenteric neurons projecting retrogradely. Dystrophic terminals in diabetic human SMG [58] were immunoreactive for neuropeptide Y (NPY), tyrosine hydroxylase, dopamine-β-hydroxylase, trkA (the cognate receptor for NGF), and p75; however, adjacent substance P, vasoactive intestinal peptide (VIP), gastrin-releasing polypeptide (GRP)/bombesin, and met-enkephalin terminals were spared. In some cases, ganglia contained increased numbers of delicate NPY processes, thought to represent axonal sprouts. This immunophenotype is consistent with origination of dystrophic axons from a subpopulation of NPY-containing noradrenergic neurons, most likely originating within the sympathetic nervous system, either intrinsic or extrinsic to the SMG, and, possibly, as locally recurrent collaterals. The neurofilaments (NF) which accumulated in diabetic and aged dystrophic sympathetic nerve terminals consisted almost exclusively of extensively phosphorylated 200-kDa NF-H epitopes [63]. Antisera directed against NF-L, NF-M, and nonphosphorylated epitopes of 200-kDa NF-H as well as MAP-2 preferentially labeled sympathetic neuronal perikarya and principal dendrites and did not label dystrophic axons. Peripherin, a 58-kDa cytoskeletal element distinct from any NF subunit which is present in subpopulations of sympathetic and DRG neurons, was colocalized with highlyphosphorylated NF-H in many dystrophic elements [63], suggesting the possibility of a shared degradative pathogenetic mechanism, rather than altered synthesis, as a target of diabetes.
Although the development of neuroaxonal dystrophy represents unambiguous and compelling pathology in the sympathetic ganglia of diabetic humans, early studies described axon loss in preganglionic sympathetic communicating (“white”)rami and greater splanchnic [57,64,65] nerves. Loss of preganglionic sympathetic innervation may, together with NAD, result in diminished numbers of normal presynaptic elements innervating principal sympathetic neurons.
Autonomic Axons in Somatic Nerves
Autonomic axons, particularly small unmyelinated axons, may be lost in somatic nerves as part of symmetrical sensorimotor neuropathy [66], which is thought to have an ischemic basis, resulting in local, distally accentuated autonomic symptoms. The loss of autonomic innervation of the vasa nervorum of somatic nerves, thought to affect blood flow to the nerve trunk, may significantly contribute to nerve ischemia described in somatic sensory polyneuropathy [67].
Diabetic Parasympathetic Nervous System
Although significant loss of vagal axons and active axonal degeneration have been described in various studies of diabetic autonomic neuropathy [68,69], the number of patients examined has typically been small. In one case of diabetic gastroparesis, dramatic axon loss in the abdominal vagus nerve was described [70]; however, a similar study failed to identify morphological abnormalities in the gastric wall or abdominal vagus nerve [71]. Immunofluorescence studies of diabetic human penis have shown preferential loss of VIP-containing axons in the corpora cavernosa [72,73].
Miscellaneous
Neuropeptide immunolocalization techniques have described decreased substance P content of human rectal mucosa in diabetic patients compared to nondiabetic controls [74]. The involvement of distal axons innervating diabetic bladder [75], skin, and penile corpora [76] has been proposed. Meissner’s and Auerbach’s plexuses in patients with diabetic diarrhea have failed to demonstrate reproducible histopathology [77], although one ultrastructural study has claimed the demonstration of marked axonal swellings within intramural ganglia [78]. PET scanning techniques have demonstrated the loss of sympathetic innervation in the distal myocardium in diabetic patients with autonomic neuropathy, although proximal segments were hyperinnervated, perhaps reflecting disorganized axonal sprouting and reinnervation [79].
Experimental Diabetic Autonomic Neuropathy
Animal models of diabetic autonomic neuropathy have been sought to provide insight into the pathogenetic mechanisms of the latter and to develop rational forms of therapy.
Sympathetic Nervous System
An unequivocal neuropathy of the alimentary tract of the streptozotocin (STZ)-diabetic and genetically diabetic BB rat and Chinese hamster has been characterized in detail [80–82]. The regular occurrence of degenerating, regenerating, and pathologically distinctive dystrophic axons has been demonstrated in: (1) preterminal axons and synapses within the prevertebral celiac and superior mesenteric sympathetic ganglia (Figs. 4.8–4.10), and (2) noradrenergic axons contained in ileal mesenteric nerves innervating the distal alimentary tract (Fig. 4.11) in rats with chronic long term STZ-induced diabetes. As in diabetic humans, NAD again developed in the prevertebral superior mesenteric and celiac ganglia but not comparably in the paravertebral superior cervical ganglia. Dystrophic swellings involved postganglionic sympathetic noradrenergic distal ileal paravascular mesenteric nerve axons and their terminals on intramural myenteric and submucosal ganglia; however, the equally lengthy noradrenergic axons which innervate the adjacent mesenteric vasculature consistently failed to develop NAD. The time course over several months of the development of NAD, its anatomical distribution (chiefly alimentary and distal), relationship to axonal length, and its response to islet cell transplantation, short- or long-term insulin therapy, aldose reductase inhibitors, and several other novel therapeutic agents (administered in a preventive or reversal mode) have been reported. A recent study [83] has demonstrated the ability of the neurotrophic substance IGF-I to reverse established neuroaxonal dystrophy in STZ-diabetic rats without correction of the metabolic severity of the diabetic state, which may reflect the known ability of IGF-I to affect axonal regeneration, collateral sprouting, or synaptic plasticity [84].
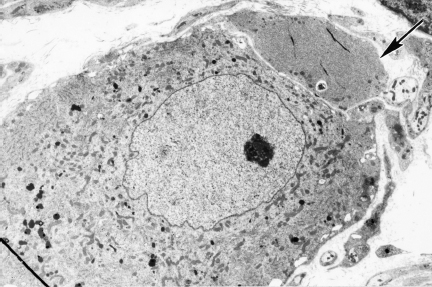
Fig. 4.8 A dystrophic axon (arrow) in the diabetic rat SMG is located within the satellite cell sheath (magnification 3000×)
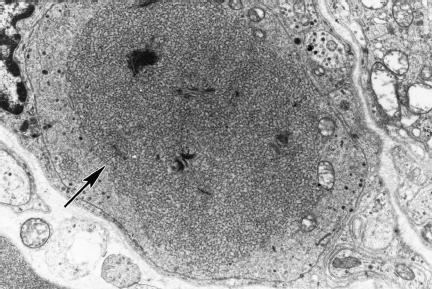
Fig. 4.9 Typically, dystrophic axons In diabetic rat SMG contain large numbers of anastomosing tubulovesicular elements (arrow; magnification 10 000×)
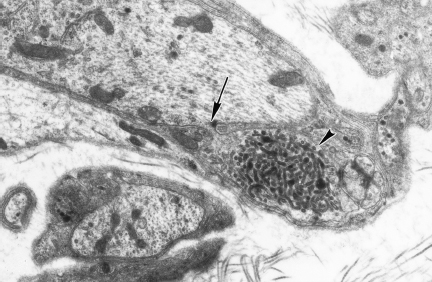
Fig. 4.10 Occasional swellings containing coarse tubulovesicular elements (arrowhead) appear to arise from projections from the adjacent perikaryon or principal dendrites, visible in this electron micrograph as a narrow cytoplasmic bridge (arrow; magnification 15 000×)
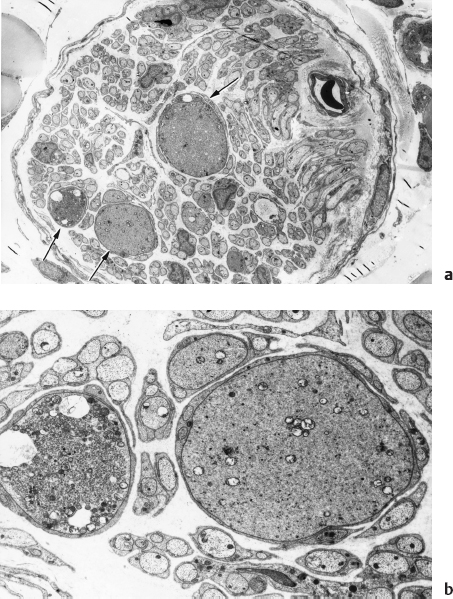
Fig. 4.11 Dystrophic axons (arrows, a), which may dominate the histologic appearance of the ileal mesenteric nerves of chronically diabetic rats, contain aggregates of tubulovesicular elements, mitochondria, and synaptic vesicles (seen better at higher magnification in b) (magnification: a 1200x; b 5000×)
Investigation of the effect of diabetes on postsynaptic dendritic structure has demonstrated dystrophic dendritic lesions (and involvement of dendritic spines in particular, Fig. 4.10) in diabetic rat prevertebral sympathetic ganglia [80,81].
The effect of diabetes on the STZ-induced diabetic rat gastrointestinal system has been further defined using electrophysiologic, immunohistologic, biochemical, and ultrastructural techniques. Degenerative changes, but not NAD, have also been described in the alimentary tract of eight-week STZ-diabetic rats, involving subpopulations of axons containing VIP [85]and calcitonin-gene-related peptide (CGRP) [86] but not substance P. Measurement of neuropeptides in diabetic rat ileum has demonstrated increased VIP and decreased substance P content [87], although changes may vary with duration of diabetes [88]. In addition, VIP and CGRP in the diabetic gut wall are not released appropriately in response to electrical stimuli [89].
Changes in neuropeptidergic and noradrenergic innervation of the diabetic rodent bowel may underlie changes in gut electrophysiology. Delayed small intestine transit time has been reported in STZ-diabetic rats [90] and in chronically diabetic Chinese hamsters [91]. Other electrophysiologic studies of the alimentary tract in experimental diabetes have also established deficiencies of cholinergic transmission [92] and muscarinic signal transduction [93], prejunctional impairment of ileal sympathetic nerve function, as well as abnormal transmucosal ionic flux apparently mediated by abnormalities in noradrenergic innervation [94].
Extra-alimentary Endorgans
Recent studies have examined the effect of diabetes on innervation of the vasa nervorum [67], heart [95] and cardiac valves, urinary bladder [96], pancreatic islets [97], and the penile corpora [98]. These studies have consistently reported decreased innervation of diabetic endorgans. However, the sympathetic innervation of the iris of long-term diabetic rats is relatively spared [99].
Parasympathetic Nervous System
Unmyelinated and myelinated axons in the vagus nerve of chronically diabetic rats [100] and Chinese hamsters [82] are reported to show axonal atrophy (but not axon loss) and regenerative changes, respectively, which may underlie changes in the variability of cardiac rhythm [100] and altered alimentary motility. Axonal atrophy and degenerative changes have also been reported in parasympathetic innervation of the diabetic rat penis, distal myenteric nerves, and urinary bladder [101,102].
Immunofluorescence studies of STZ-diabetic rat penis have shown preferential loss of VIP-containing axons in the corpora cavernosa [73] and selective degeneration of nitrergic nerves [103].
Pathogenetic Mechanisms
The terminal aspects of autonomic axons appear to be preferentially targeted in diabetes. Ganglionic neuroaxonal dystrophy may represent an abnormal outcome of synaptic turnover, which may normally sub-serve synaptic plasticity, or the synaptic detachment/reattachment process that follows postganglionic sympathetic axotomy [104]. Other possible pathogenetic mechanisms have been previously described in detail [105].
Pathobiochemistry and Pathophysiology
 Glycemic Control
Glycemic Control
D. Ziegler
Introduction
It was as early as in 1864 when Marchal de Calvi established that neurologic symptoms reflect the consequence rather than the cause of diabetes mellitus [106], However, one of the most intriguing questions in clinical research in diabetes during the past decades was whether long-term near-normoglycemia may retard or improve the chronic diabetic complications, including diabetic neuropathy [107,108]. The recent publications of the two largest and longest studies in the history of diabetes research, the Diabetes Control and Complications Trial (DCCT), conducted in type 1 diabetic patients, and the United Kingdom Prospective Diabetes Study (UKPDS), performed in type 2 diabetic patients, have been interpreted as providing evidence of the benefit of intensive diabetes therapy on the development and progression of the chronic diabetic complications [109–111]. However, while in both studies the effects of improved glycemic control on the microvascular endpoints were unanimously considered as being favorable [107,112], the effects on macrovascular endpoints in the UKPDS have also been interpreted as showing a clinically important benefit on macrovascular endpoints only in patients treated with metformin, but not those treated with sulfonylureas or insulin. Because metformin provided blood glucose levels similar to those of sulfonylureas or insulin, the benefit from metformin appeared to be independent of its blood-glucose-lowering effect [113]. Moreover, in both studies microvascular or macrovascular rather than neuropathic endpoints were used as the primary outcome measures.
Numerous previous short-term studies have shown that neuropathic symptoms or abnormal nerve function tests occurring during periods of metabolic derangement can be ameliorated within several days or weeks following improvement of blood glucose control [114–122]. However, possible long-term effects have been difficult to study due to the following problems: (1) the progression of diabetic polyneuropathy is relatively slow, so that expected changes may take place over several years, (2) the various nerve fiber populations might be affected at different rates, (3) minor changes may not be detected due to a low reproducibility of some methods, (4) glycemic control or the risk factor profile may fluctuate over time, and (5) even with the modern intensive diabetes therapy regimens, long-term near-normoglycemia is difficult to achieve in some patients. These problems may in part account for the conflicting findings in the earlier reports, with some showing improvement of peripheral nerve function [123–125], while others have failed to demonstrate any changes [126,127].
Rapidly Reversible Nerve Dysfunction After Correction of Metabolic Derangement
Untreated, newly diagnosed type 1 diabetic patients show slight reduction in motor nerve conduction velocity (NCV), which has been shown to improve significantly as soon as one week following the elimination of ketosis and hyperglycemia by insulin treatment [116–118]. Similarly, other findings of neural dysfunction, such as increased resistance to ischemia in the diabetic nerve [118] or impaired retinal neurophysiologic function [119] were rapidly reversible within one to three weeks of improved glycemic control after the diagnosis of type 1 diabetes. In hyperglycemic patients with various durations of diabetes, who were treated with an artificial endocrine pancreas, H-reflex conduction velocity or motor and sensory conduction increased significantly during two or three days of normoglycemia [120,121]. Metabolic derangement resulting in diabetic ketoacidosis is often associated with nerve conduction slowing which has been shown to be reversible following insulin treatment for three months or less [122].
The aforementioned studies suggest that the acutely reversible changes in nerve function observed after improvement in glycemic control are attributable to functional rather than structural alterations in the diabetic nerve during episodes of metabolic derangement. Experimental studies have shown a marked reduction of the compound nerve action potential in isolated dorsal rat spinal roots incubated in 25-mM extracellular glucose and transiently exposed to hypoxia. This electrophysiologic alteration appeared to be caused by acidosis, because it was prevented when bicarbonate-containing solutions were used [128].
Although the rapidly reversible abnormalities in nerve function are related to restoring near-normoglycemia, acute painful neuropathy associated with the initiation of tight glycemic control has been reported in some patients [129]. Caravati [130] first described this rare phenomenon, which he called “insulin neuritis,” in 1933 (see Chapter 5, page 306–309). This effect has been observed in poorly controlled patients with markedly raised HbA1 levels and occurred within several weeks of lowering of blood glucose by intensive insulin treatment but without evidence for frequent hypoglycemic episodes. Continuation of insulin treatment and maintenance of good glycemic control leads to a recovery from the painful symptoms after periods of up to six months. Sural nerve biopsy in one case during the acute phase revealed predominant small-fiber loss and regenerating axon sprouts [129].
Role of Intensive Diabetes Therapy in Treatment and Prevention of Diabetic Neuropathy
Earlier Small Trials in Type 1 Diabetic Patients
Earlier uncontrolled short-term studies including relatively small numbers of patients with diabetic neuropathy have reported that neuropathic symptoms or abnormal nerve function tests seen during hyperglycemic conditions may be more or less ameliorated following improvement of glycemic control [131–134]. Previous randomized controlled studies have assessed the influence of improved glycemic control on peripheral nerve function for periods of up to two years. However, several shortcomings are apparent in these studies: (1) two studies have used retinopathy as the primary selection criterion for entry and provided no information as to the prevalence and severity of clinical neuropathy [123,127], (2) only one study used reliable and clinically meaningful criteria for the diagnosis of neuropathy [126], (3) three studies did not measure nerve conduction [123,125,127], which is thought to be the most objective, sensitive, and reliable test in the evaluation of diabetic neuropathy [135], and (4) intensified insulin treatment did not lower the elevated HbA1 values to the normal range. In addition, in most of these studies the differences in mean HbA1 between the conventionally and intensively treated patients were relatively rather small to result in meaningful differences in peripheral nerve function. According to Dyck and O’Brien [136], the following degrees of changes in motor and sensory NCV that are associated with a change in the Neuropathy Impairment Score (NIS) of two points can be regarded as meaningful in controlled clinical trials: median motor NCV: 2.5 m/s, ulnar motor NCV: 4.6 m/s, peroneal motor NCV: 2.2 m/s, median sensory NCV: 1.9 m/s, and sural sensory NCV: 5.6 m/s. A change in NIS of two points corresponds to, e.g., bilateral change in dorsiflexor muscle strength of 25%, or change in ankle reflexes, or pin-prick perception from normal to decreased and vice versa.
Ziegler et al. [137] conducted a prospective study in 55 initially poorly controlled type 1 diabetic patients who were treated with continuous subcutaneous insulin infusion (CSII) or intensive conventional therapy (ICT) for four years. Patients were divided into three groups according to their mean HbA1 levels during the study. Group 1 (n = 19) had mean HbA1 during months 3-48 in the normal range of less than 7.8% (near-normoglycemic control), group 2 (n = 18) showed moderately elevated mean HbA] between 7.8% and 8.5% (satisfactory control), and group 3 (n = 18) had clearly elevated mean HbA1 of 8.6% or above (poor control). In the three groups studied, the changes in median and peroneal motor NCV over baseline as well as median and ulnar sensory NCV after four years were inversely related to the mean HbA1 levels of months 3-48 (P<0.05). No significant associations with mean HbA1 were noted for ulnar motor NCV, sural sensory NCV, and heart rate variability (HRV) as an index of autonomic dysfunction. Thus, near-normoglycemia maintained for four years in type 1 diabetic patients was associated with an increase in NCV in the upper limbs but not sensory NCV in the lower limbs and HRV. These results indicate that the susceptibility of different nerve fiber populations to long-term improvement in blood glucose control may be variable.
Long-Term Trials in Type 1 Diabetic Patients
Three pivotal long-term prospective studies that included type 1 diabetic patients either with mild retinopathy or without evidence of diabetic complications have been published (Table 4.1). The results of the Stockholm Diabetes Intervention Study (SDIS) over 10 years [138], the Oslo Study over eight years [139], and the DCCT over five years [140] demonstrate that long-term near-normoglycemia retards the deterioration in motor and sensory NCV. In the DCCT, intensive insulin therapy reduced the appearance of nerve conduction deficits after five years by approximately 50%. The risk for the development of clinical neuropathy was reduced by 64% within five years (5% vs 13% for the intensive therapy [IT] vs the conventional therapy [CT] group) [109]. Most attributes of nerve conduction remained stable or showed modest improvement in patients on IT, whereas they generally deteriorated in those on CT. Among patients in the primary prevention cohort (for retinopathy) without neuropathy at baseline, the IT group had significantly higher NCVs at five years compared with the CT group, the most prominent difference being noted for the peroneal motor NCV, which was 4.1 m/s faster [140]. A comparable effect was observed in the subgroup of patients with possible or definite neuropathy at baseline and in the secondary intervention cohort. Thus, the magnitude of treatment effect was relatively independent of the presence or absence of clinical neuropathy at baseline. Abnormal R-R interval variation at deep breathing as a measure of cardiovascular autonomic neuropathy (CAN) was significantly more frequent in the CT group than the IT group (14.8% vs 7.6%) in the secondary intervention cohort at 5-6 years [141]. The corresponding percentages for any abnormality among R-R interval variation, Valsalva ratio, and postural testing in the secondary intervention cohort at 5-6 years were 16.2% and 8.2%, respectively. These differences were considerably smaller and did not reach statistical significance in the primary prevention cohort. Overall, less than 3% of the DCCT subjects reported symptoms consistent with autonomic dysfunction. Thus, intensive therapy can slow the progression and development of autonomic dysfunction in type 1 diabetic patients with retinopathy.
The Epidemiology of Diabetes Interventions and Complications (EDIC) study, a long-term observational continuation of the DCCT in which the CT patients were offered IT, showed that the reduction in the risk of progressive retinopathy and nephropathy resulting from IT persists for four years, despite a narrowing in the difference in mean HbA1c between the groups, which decreased from 9.1% to 8.2% in the original CT group and increased from 7.2.% to 7.9% in the IT group [142]. However, no data were reported for neuropathy.
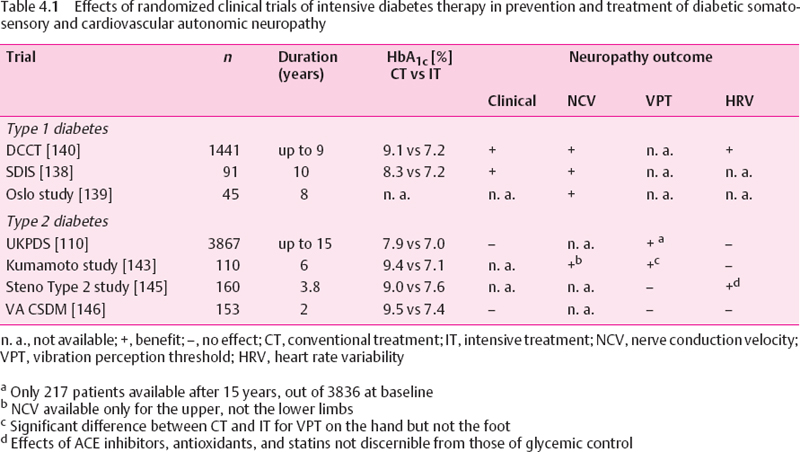
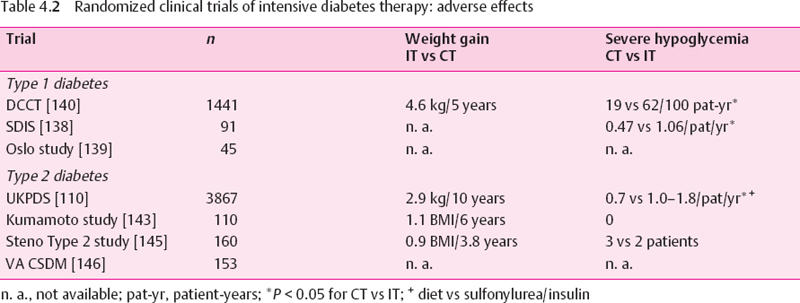
In the Oslo Study, a difference in HbA1 of 1% was associated with changes in peroneal motor NCV of 1.2 m/s, in tibial motor NCV of 1.3 m/s, and in sural sensory NCV of 1.4 m/s during the eight-year follow-up period [139], In the SD1S the mean differences in NCV between the IT and CT groups after 10 years were 5.1 m/s for the peroneal nerve. 6.0 m/s for the tibial nerve, and 8.9 m/s for the sural nerve. Pin-prick sensitivity deteriorated significantly in the CT vs the IT groups, and the rates of neuropathic symptoms were 14% vs 32% after 10 years for IT vs CT, respectively [138], However, these studies do not provide information about the reversibility of established nerve conduction deficits. Fur-thermore, in the Oslo Study the original randomization to the three treatment groups was abolished, and the patients were retrospectively allocated to new groups according to their mean HbA1 over eight years. A certain degree of deterioration and development of deficits in NCV was also observed in the well-controlled groups, suggesting that, using the current methods of intensive insulin therapy, complete prevention of neuropathy is difficult to achieve. Moreover, in the DCCT, intensive therapy was associated with a three-fold increased risk of severe hypoglycemia and mean weight gain of 4.6 kg within five years. In the SDIS, a two-fold increased risk of severe hypoglycemia was observed for intensive therapy (Table 4.2).
Long-term Trials In Type 2 Diabetic Patients
The UKPDS showed a lower rate of impaired vibration perception threshold (VPT>25 V) after 15 years for IT with a sulfonylurea or insulin vs CT with diet (31% vs 52%). However, only 217 patients were available for assessment of VPT after 15 years, out of 3836 available at baseline. Thus, a bias due to the small sample size may have influenced this result. Moreover, the only additional time point at which VPT reached a significant difference between IT and CT was the nine-year follow-up, whereas the results after three, six, and 12 years did not differ between the groups. Likewise, the rates of absent knee and ankle reflexes as well as the heart rate responses to deep breathing did not differ between the groups. IT was associated with an increased risk of weight gain and hypoglycemia [110] (Table 4.2).
In the Kumamoto Study of insulin-treated type 2 diabetic patients, after six years NCV in the median motor and sensory nerves was significantly slower in patients given conventional insulin injection therapy (CT) compared with those given multiple insulin injections (IT), but unfortunately NCV was not measured in the lower limbs, where it would be more likely to reflect an effect on polyneuropathy. This is particularly important in view of the fact that VPT in the upper but not the lower limbs was significantly improved in IT as compared with CT. Similar to the findings of the UKPDS. HRV at rest and during deep breathing and posture-related change in blood pressure did not differ between the groups after six years [143]. The 10-year follow-up of the Kumamoto Study showed a relative risk reduction in the progression of clinical neuropathy by 64% in the IT group compared to the CT group. Moreover, IT prolonged the period in which patients were free of clinical neuropathy by 2.2 years, and was more cost-effective, mainly because of reduced costs for management of diabetic complications [144].
In the Steno type 2 Study, intensified multifactorial intervention including the use of intensive diabetes treatment, ACE inhibitors, antioxidants, statins, aspirin, and smoking cessation in patients with microalbuminuria had no effect on the progression of polyneuropathy after 3.8 years. By contrast, a positive effect of this approach was seen on HRV. It cannot be deduced from this study that this effect was due to improved glycemic control, because any of the other interventions, particularly the administration of ACE inhibitors and antioxidants, or even smoking cessation, may have been responsible for this result [145].
In the Veterans Administration Cooperative Study on type 2 diabetes mellitus (VA CSDM), no significant effect of IT (four-step plan of multiple insulin injections) compared to CT (one morning insulin injection per day) on peripheral neuropathy, abnormal Valsalva ratio and/or R-R interval variation, or erectile dysfunction could be demonstrated after two years, despite a difference in HbA1c between the groups comparable with those in the above studies [146].
In conclusion, these trials have shown heterogeneous effects of intensive diabetes therapy on the progression of distal symmetric polyneuropathy and autonomic neuropathy. It cannot be concluded unequivocally from these results that improved glycemic control prevents the development or retards the progression of polyneuropathy in type 2 diabetic patients treated for periods of 2-15 years. In the UKPDS intensive therapy was associated with an increased risk of weight gain and mild or severe hypoglycemia.
Pancreas Transplantation in Type 1 Diabetic Patients
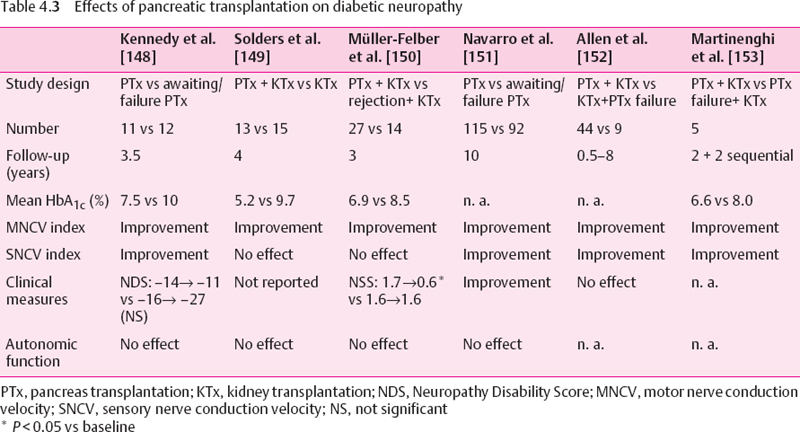
Pancreas transplantation is the most effective method of achieving long-term normoglycemia in type 1 diabetic patients, but is usually limited to patients with end-stage diabetic nephropathy in combination with a renal graft. Other indications have been questioned [147]. Several long-term studies in patients with established diabetic polyneuropathy who underwent successful pancreatic transplantation have been published (Table 4.3). Kennedy et al. [148] have shown that 42 months after transplantation the neuropathy was only slightly improved, but a significant difference was seen in the mean motor and sensory nerve conduction in the transplanted group compared with a control group who did not have a functioning graft after 42 months. Improvement was more pronounced when only mild dysfunction was present initially.
Solders et al. [149] have demonstrated beneficial effects of combined pancreatic and renal transplantation on NCV after four years of normoglycemia. The initial improvement in motor NCV observed in these patients was also noted in diabetic patients receiving a renal transplant only, and was most likely due to the elimination of uremia. However, further improvement was seen only in the euglycemic pancreas graft recipients. Muller-Felber et al. [150] have shown modest improvement in neuropathic symptoms and increase in motor but not sensory NCV after three years in patients who underwent successful pancreas and kidney transplantation as compared to those with early pancreas rejection and functioning kidney graft. None of the beneficial effects described in these three studies were demonstrable after two years, and none of these studies could demonstrate an effect on HRV as an index of cardiovascular autonomic dysfunction, while the effects on clinical measures were variable (Table 4.3). These findings are confirmed in the more recent studies shown in the last three columns of Table 4.3 [151–153]. Thus, periods of normoglycemia of more than two years following pancreas transplantation retard the further progression of deficits in motor and sensory NCV, but no such clear effect is noted for neuropathic symptoms and deficits, while no beneficial effect is seen on cardiovascular autonomic dysfunction. The reasons for this divergent effect are not known but may be due to a different susceptibility of the autonomic nerves to metabolic changes or to methodological factors.
Is There a Glycemic Threshold for the Risk of Diabetic Complications?
Two retrospective studies suggested the existence of a glycemic threshold at a HbA1c level of approximately 8% for microalbuminuria and retinopathy, below which there is no further reduction in risk [154,155]. This would imply that improving glycemic control below this level is unnecessary, thereby potentially reducing the risk of hypoglycemia in some type 1 diabetic patients [156]. In contrast, the DCCT could not identify such a HbA1c threshold. The risks of retinopathy progression and of developing microalbuminuria and neuropathy were found to be continuous but nonlinear over the entire range of HbA1c values. As HbA1c was reduced below 8% there were continuing relative reductions in the risk of diabetic complications, whereas the rate of increase in the risk of hypoglycemia was slower [157,158]. Likewise, in the Pittsburgh Epidemiology of Diabetes Complications Study no definitive threshold was found after six years for any complication in type 1 diabetic subjects [159].
Conclusions
1. The large randomized long-term clinical trials such as the DCCT and UKPDS were not designed to evaluate the effects of intensive diabetes therapy on diabetic polyneuropathy, but rather to study the influence of such treatment on the development and progression of the chronic diabetic complications. Thus, only a minority of the patients enrolled in these studies had symptomatic polyneuropathy at entry. In type 1 diabetic patients these studies show that intensive diabetes therapy retards but does not completely prevent the development of polyneuropathy and autonomic neuropathy. In contrast, in type 2 diabetic patients, who represent the vast majority of people with diabetes, the results were variable. Intensive diabetes therapy either had no effect or only partially slowed the progression of polyneuropathy, and the effect on autonomic neuropathy was largely lacking. Moreover, improved glycemic control was achieved at the expense of increased risk of hypoglycemia and weight gain.
2. Only a few small studies have evaluated the effects of intensive diabetes therapy on established polyneuropathy in type 1 diabetic patients. They indicate that improved glycemic control may improve some aspects of diabetic neuropathy, but imperfect study designs and methodology hamper the validity of most of these small trials. At more advanced stages improvement is still possible for some measures of nerve function such as motor NCV, but is less likely for autonomic dysfunction. This may be due to the fact that true normoglycemia could not be achieved in many patients. No large, randomized, controlled trial has been performed specifically to show favorable effects of intensive diabetes therapy on diabetic polyneuropathy.
3. In type 1 diabetic patients with the most advanced stages of peripheral neuropathy, progression of nerve conduction deficits is halted after three to four years of normoglycemia following pancreas transplantation, but no effect is seen in autonomic neuropathy. However, successful pancreas transplantation results in long-term normoglycemia. Hence, the effect on nerve function that can be achieved with this method cannot be extrapolated to the widely used current methods of intensive diabetes therapy, since for various reasons the majority of diabetic patients in whom these methods are used do not achieve sustained normoglycemia.
4. Although observational studies suggested a glycemic threshold for the development and progression of long-term complications in type 1 diabetes, the DCCT data do not support such an assumption. Thus, attempts to achieve optimal glycemic control should not aim at a particular HbA1c threshold within the diabetic range, but should follow “the goal of achieving normal glycemia as early as possible in as many IDDM patients as is safely possible” [158].
 Metabolic Alterations in Experimental Models
Metabolic Alterations in Experimental Models
A.A.F. Sima and C.R. Pierson
Introduction
Glycemic control is the treatment goal of managing diabetic patients. The Diabetes Control and Complications Trial (DCCT) demonstrated that intensive insulin treatment to control serum glucose reduces the onset of clinical neuropathy at five years by 57% [109]. Epidemiologic data show that only 50% of diabetic subjects experience clinical manifestations of diabetic neuropathy [160], suggesting that other metabolic and genetic factors may be at play. However, hyperglycemia is probably the most important initiator of diabetic neuropathy, by activating the polyol pathway, affecting nonenzymatic glycation, nerve blood flow, and leading to the generation of reactive oxygen species (ROS). All of these metabolic factors contribute to the development of diabetic neuropathy and will be dealt with in more detail elsewhere in this volume. The fact that, despite intensive insulin therapy and near-normal glycemic control, diabetic neuropathy still occurred in the DCCT cohort suggests that additional factors may be of pathogenic significance in diabetic neuropathy. Such factors include insulin and/or C-peptide deficiencies in type 1 diabetes and hyperinsulinemia in type 2 diabetes and genetic susceptibility [161,162]. Support for a role of insulin and/or C-peptide deficiencies stems from the differences in severity of the neuropathy encountered in type 1 and type 2 diabetes. Studies have demonstrated that in human diabetic neuropathy, type 1 diabetes is a prominent risk factor for the severity of diabetic neuropathy [163]. More severe structural and functional alterations are evident in animal models of type 1 neuropathy than in type 2 neuropathy despite comparable levels of hyperglycemia [164–167]. Insulin and C-peptide deficiencies characterize type 1 diabetes, whereas these hormones are unchanged or increased in type 2 diabetes. Therefore, do insulin and C-peptide possess neuroprotective effects, and could their deficiencies in type 1 diabetes account for its association with more severe diabetic neuropathy than that of type 2 diabetes? In this section we will explore the role of both hyperglycemia and hypoglycemia, and the potential contribution of insulin and C-peptide deficiencies to the development of diabetic neuropathy.
How the metabolic alterations of diabetes contribute to the pathogenesis of neuropathy has been the subject of extensive research during the last 20-30 years. Available data indicate that the metabolic causes of diabetic neuropathy are multifactorial, interrelated, and mutually perpetuating, and have until recently been ascribed solely to hyperglycemia [168]. The metabolic alterations involved include the activation of the polyol pathway, nonenzymatic glycation, impaired nerve blood flow, and generation of ROS. Each of these mechanisms appears to be most prominent during a certain phase of diabetic neuropathy, but they are interrelated and mutually perpetuating (Fig. 4.12). The accumulation of sorbitol from increased polyol pathway flux is an early event in experimental [169] and human diabetic neuropathy [170] and increased sorbitol content is not evident in chronically diabetic human nerve [171]. Studies in BB/W rats show that sorbitol accumulation, myoinositol depletion, and impaired Na+, K+-ATPase activity wane as the neuropathy progresses and additional metabolic pathways become more prominent [169]. By contrast, the reduction in the expression of neurotrophic factors [172] develops later in the course of the disease.
The Polyol Pathway and the Na+, K+-ATPase Defect
Hyperglycemia enhances flux through the polyol pathway resulting in glucose being converted by the high-Km enzyme aldose reductase into sorbitol and to fructose by sorbitol dehydrogenase [173]. Immunohisto-chemical studies have localized polyol pathway enzymes to a variety of sites including the nodes of Ranvier of myelinated fibers and the endoneurial vasculature [174,175]. Sorbitol is an organic osmolyte and its accumulation leads to a compensatory depletion of other osmolytes such as myo-inositol and taurine [176]. As an example of the intricacy of these metabolic alterations, taurine is also an antioxidant and promoter of nerve regeneration, and hence contributes to these pathogenic processes [177,178]. The loss of myo-inositol leads to alterations in phosphoinositide metabolism, with decreased availability of diacylglycerol [173], which contributes to the reduction in neural Na+, K+-ATPase activity. Perturbed Na+, K+-ATPase activity leads to intra-axonal Na+ accumulation and decreased nodal Na+ equilibrium potential, resulting in the early reversible nerve conduction velocity (NCV) defect [179–181]. These relationships account for the beneficial effects of aldose reductase inhibition and myo-inositol supplementation on Na+.K+-ATPase activity and NCV in experimental models of acute diabetes [179].
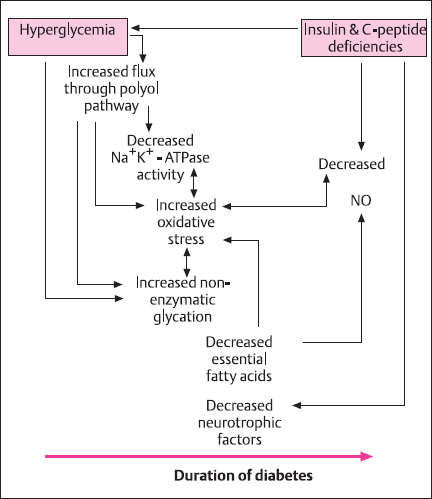
Fig. 4.12 Metabolic alterations caused by hyperglycemia and/or insulin and C-peptide deficiencies. The individual metabolic abnormalities occur sequentially and are often mutually perpetuating
Na+, K+-ATPase activity can be modulated by means other than the polyol pathway, such as impaired nitric oxide synthesis [173], pseudohypoxia [182], and prostacyclin deficiency [183]. The normalizing effect on neural Na+, K+-ATPase activity can be achieved by prostacyclin analogues [184], acetyl-L-carnitine [185], and C-peptide [164,167]. The Na+,K+-ATPase deficit is therefore linked to multiple metabolic alterations, not just those resulting from altered flux through the polyol pathway. Acetyl-L-carnitine, which has no effect on the polyol pathway, has been shown to correct nerve Na+, K+-ATPase activity [169,186], blood flow [187,188], levels of vasoactive prostaglandins, and NCV [169]. C-peptide also normalizes Na+, K+-ATPase activity believed to occur via a Ca2+-mediated pathway [164,189]. Taken together, these data indicate that the alterations in Na+, K+-ATPase activity are the result of several metabolic alterations.
The activation of the polyol pathway by hyperglycemia leads to increased oxidative stress, and aldose reductase inhibitor (ARI) treatment has been shown to decrease oxidative stress in the sciatic nerve of diabetic rats [190]. NADPH is used as a cofactor not only for aldose reductase, but also for the glutathione cycle and nitric oxide synthase (NOS). Therefore, activation of the polyol pathway due to hyperglycemia depletes NADPH, limiting the amount of NADPH available for the recycling of reduced glutathione (GSH) from its oxidized form, GSSC, and nitric oxide synthesis. In the second step of the polyol pathway, sorbitol is oxidized to fructose, which increases the NADH/NAD+ and the lactate/pyruvate ratio. This state has been termed “pseudohypoxia” by Williamson et al. [182] and was improved by sorbitol dehydrogenase inhibition [191]. However. Cameron et al. [192] and Schmidt et al. [193] were unable to reproduce these results.
The relative lack of structural changes in the peripheral nerve such as demyelination or fiber degeneration, coincident with the acute slowing of NCV, suggests underlying biochemical mechanisms [194]. The association between acute metabolic alterations, slowing of NCV, and decreased Na+, K+-ATPase activity is confirmed by direct measurements of Na+, K+-ATPase activity accompanied by decreased NCV. Structurally, the diminished Na+, K+-ATPase activity manifests as early paranodal swellings due to intra-axonal accumulation of Na+ [181,194]. These changes are correctable by insulin treatment [181]. myo-inositol supplementation [179,180], ARI treatment [180], acetyl-L-carnitine administration [169], and C-peptide treatment [164,167].
Experimental models of type 1 diabetic neuropathy demonstrate that increased flux through the polyol pathway is a critical component in the early pathogenesis of diabetic neuropathy. Activation of the polyol pathway also impacts secondarily by promoting generation of ROS and nonenzymatic glycation and perturbing nitric oxide synthesis and blood flow [173]. Despite the prominence of the early polyol pathway activation in human and rat diabetic neuropathy, it may not be an absolute requirement for the development of diabetic neuropathy. Diabetic mice, which show no or low polyol pathway activity, still exhibit functional and structural diabetic neuropathy similar to that seen in rat models [195,196].
Role of Nonenzymatic Glycation
Nonenzymatic glycation is enhanced in the peripheral nerve in both experimental animal models and human diabetic subjects [197,198]. In this process, reducing monosaccharides combine with free amino groups on proteins or other cellular molecules forming early reversible products, known as Schiff bases or ketamines or Amadori adducts. In the Wolff pathway, glycoxidative products form and contribute to cellular oxidative stress [199]. Advanced glycation end products (AGE) form through critical intermediates such as 3-deoxy-glucosone (3DG) from fructose-Iysine and glyoxal, and methylgiyoxal (MGO), from Amadori compounds, Schiff bases, or by the direct oxidation of monosaccharides [200–202]. Subsequent chemical rearrangements, dehydration, fragmentation, and cross-linking occur, forming irreversible AGEs.
A number of compounds participate in the glycation process, including glucose, glucose-6-phosphate, galactose, pentose phosphate pathway compounds, various trioses, and fructose generated via the polyol pathway [203]. Fructose is a potent glycator and its activity far exceeds that of glucose [203]. Fructose can be metabolized to fructose-6-phosphate and triose phosphate and these intermediates are sources of 3DG and MGO, both of which induce oxidative stress via cross-link formation [204]. Interestingly, the activity of sorbitol dehydrogenase decreases following its glycation [205]. Hence accumulation of fructose from the increased polyol pathway flux may contribute to inhibition of sorbitol dehydrogenase, providing a negative feedback mechanism [168]. Aldose reductase inhibition in humans and experimental animals results in a reduction of glycated protein content in various tissues; however, the levels in peripheral nerve have not been ascertained [206].
AGEs accumulate in specific tissue constituents of peripheral nerve including certain myelin components such as P0, myelin basic protein, and proteolipid protein [197,207], This raises the possibility that AGE-modified myelin proteins may contribute to segmental demyelination in diabetes via their recognition by AGE-specific receptors on macrophages, resulting in myelin breakdown and phagocytosis [198]. The receptor for AGE (RAGE) has been characterized and is considered a member of the immunoglobulin superfamily of cell adhesion molecules, with considerable homology to the neural cell adhesion molecule (NCAM) [208,209], The RAGE-ligand interaction generates intracellular oxidative stress and activates NF-kB, a redox sensitive transcription factor [210,211].
Axonal cytoskeletal proteins such as tubulin, neurofilament, and actin are glycated, resulting in abnormalities in axonal structure, slow transport, atrophy, and degeneration [198,212], It is suggested that glycation of these cytoskeletal components hampers their polymerization and phosphorylation and, ultimately, their exportation from the perikaryon. Glycation of extracellular matrix proteins may compromise nerve regeneration ability in diabetic neuropathy. Glycation of laminin, a molecule that serves as guidance for the axonal growth cone, may impact upon both initiation and sustaining of nerve regeneration [213].
AGEs are capable of generating cellular oxidative stress. Two compounds, Nε-[carboxymethyl]-lysine (CML) and pentosidine, result from glycation and oxidation reactions and are considered markers of glycative and oxidative stress [214]. CML immmunolocalizes to perineural basal laminae, axons, Schwann cells, and endoneurial microvessels in human nerve. The immunoreactivity increases significantly with diabetes and correlates with fiber loss [215]. Glycation may perpetuate oxidative stress by inactivation of protective enzymes such as Cu-Zn-superoxide dismutase [216]. AGEs binding to RAGE on macrophages generate oxidative stress as GSH and vitamin C depletion results, which is accompanied by nuclear factor-KB (NF-kB) induction [217], Treatment with α-lipoic acid, an antioxidant, prevents the induction of NF-kB in this scenario [217].
Aminoguanidine inhibits the formation of AGE by preventing cross-linking, and has a beneficial effect on neuropathy, nephropathy, and retinopathy in diabetic rats. In peripheral nerve aminoguanidine improves NCV and morphometric parameters after long-term treatment, as its beneficial effects become apparent only after a lag period. Nerve blood flow improves in diabetic animals after aminoguanidine treatment; however, no effect on free radical activity could be detected [218,219]. When administered short-term, aminoguanidine improves NCV and the Na+, K+-ATPase defect; however, long-term administration is required before structural improvements of the endoneurial vasculature are evident [220,221].
Role of Oxidative Stress and Alterations in Blood Flow
Diabetes is a state of increased oxidative stress characterized by both an increased production of ROS and impaired ability to scavenge ROS; however, the precise mechanisms responsible for these phenomena are not completely understood and are probably multifactorial [214,222,223]. Superoxide, hydrogen peroxide, and hydroxyl radicals are toxic products derived from the chemical reduction of molecular oxygen via the Haber-Weiss reaction [224]. ROS promote the autoxidation of cellular molecules such as glucose, thiols, and catecholamines, and the rate of this reaction can be accelerated by transition metals such as copper and iron [224]. Polyunsaturated fatty acids are especially vulnerable to peroxidation by ROS, and this reaction can initiate an autocatalytic chain reaction which peroxidizes other fatty acids [225]. Other targets of ROS peroxidation include proteins and DNA.
Ceils are not defenseless against ROS; superoxide dismutase scavenges superoxide in both cytoplasm and mitochondria. Glutathione peroxidase destroys lipid hydroperoxides and hydrogen peroxide. Most of the scavenging mechanisms are not due to enzymatic activity but rather to a variety of compounds that interact with ROS. These compounds can be placed into two groups based upon their chemistry, those that are hydrophobic, such as vitamin E (α-tocopherol), ubiquinone, and carotenoids, and can inactivate hydrophobic ROS, and those that are hydrophilic and capable of inactivating hydrophilic ROS, such as ascorbic acid and cystine [225].
The vascular system bears a considerable burden from the pro-oxidant activity in the diabetic state. Oxidative stress underlies the propensity to develop microvascular complications and atherosclerosis. The microvascular complications are pertinent to the development of neuropathy in diabetes and include alterations in the function of endothelial cells and vascular reactivity, which in turn leads to impaired endoneural blood flow and oxygenation.
Endothelial cell function and gene expression are altered by the pro-oxidant state. The resulting altered gene expression favors endothelial-mediated vasoconstriction, which compromises nerve blood flow. Serum levels of angiotensin-converting enzyme are increased in both diabetic human and rat models [22], and the renin-angiotensin system is up-regulated in the vasa nervorum [227]. The increase in serum angiotensin-converting enzyme is prevented by administration of the antioxidant probucol in experimental models [185]. Oxidative stress leads to an increase in the expression of endothelin-1 (ET-1), a potent vasoconstrictor. NF-kB probably serves as the mediator of ET-1 transcription as NF-kB consensus binding sites are present in the ET-1 promoter [228]. BQ123 and BM5 182874 are ETA receptor antagonists, and when administered to experimental animal models they restore blood flow and NCV [229]. Basentan is an ETA ETB antagonist, and when it is used to treat diabetic rats a smaller effect on nerve blood flow and NCV is observed [230]. This is probably due to the fact that ETB stimulation produces nitric oxide (NO) and prostacyclin, and the activity of these vasodilators probably counters the vasoconstrictive activity of ETA stimulation [229]. Superoxide dismutase generates peroxynitrite from NO, which is toxic to endothelial cells, and further illustrates the need for a proper balance between antioxidative factors and oxidative stress [231]. Increased leukocyte adhesion to endothelial cells also results from NF-kB activity, and this may explain a component of the increased propensity to thrombus formation in diabetics [232]. Antioxidant therapy with α-tocopherol and probucol prevents leukocyte adhesion [233].
Nonspecific and β-specific protein kinase C (PKC) inhibitors normalize NCV and sciatic nerve blood flow in the streptozotocin (STZ)-induced diabetic rat [234,235]. α-lipoic acid is an antioxidant and has been shown to correct myo-inositol depletion, perhaps by augmenting Na+, K+-ATPase activity or increasing energy production by normalizing blood nerve blood flow [236]. Vasodilators such as niceritol and β-specific PKC inhibitors have comparable effects, which may be exerted by similar mechanisms [237].
Acute hyperglycemia alters endotheliai-mediated vascular relaxation. In aortic rings incubated in high-glucose medium a reduction in acetylcholine-mediated endothelial relaxation is elicited, and this change can be prevented by probucol or superoxide dismutase administration [238,239]. Acetylcysteine, another antioxidant, has also been shown to improve nerve blood flow and NCV [240]. Acetylcysteine and α-tocopherol mitigate axonal atrophy and enhance nerve fiber regeneration in experimental diabetic neuropathy, further supporting the role of oxidative stress [241,242].
Transition metals are probably important for the generation of oxidative stress in diabetic peripheral nerve. Transition metals catalyze the autoxidation of glucose, fructose, and other monosaccharides and AGEs to ROS. Diabetic rats treated with metal chelators show a correction of nerve blood flow and NCV [225].
Assessment of the expression and activity of nitric oxide synthase (NOS), which produces the critical vasodilator NO, in 12-month STZ-diabetic rat dorsal root ganglia (DRGs) revealed increased enzyme activity but no increase in NOS expression. In the same study, at two months of diabetes endothelial NOS (eNOS) immunoreactivity acquired a novel pattern, with perineural staining of sciatic nerve and capsular staining of DRGs, which was not evident in long-standing diabetes. Therefore, since increases in NO production were not associated with increases in NOS expression, it may be that enhanced efficiency of NOS enzymatic activity, rather than increased expression, accounts for the increase of NOS activity in diabetes [243]. One intriguing finding is that C-peptide mediates vasodilatation of muscle vascular beds by a mechanism that incorporates NO [244]. The potential pathogenic roles of NO and C-peptide in diabetic neuropathy remain unexplored.
Role of Essential Fatty Acids
Diabetes leads to alterations in the levels of essential fatty acids. The synthesis of prostanoids, which possess critical vasoactive properties, is markedly decreased in diabetes [225]. This can be attributed to impaired desaturation at the Δ-6 step in the synthetic pathway and additional deficits in ω-6 essential fatty acids. Hepatocyte desaturation activity is likely due to hyperglycemia, hypoinsulinemia, and oxidative stress [245]. Deficient Δ-6 desaturation reduces tissue and plasma levels of γ;-Iinoleic acid and arachidonic acid, so a dearth of products normally generated by cyclo-oxygenase catalytic activity results. These products include prostaglandins with vasodilatory and antiplatelet effects [226], so increased vascular tone and diminished nerve blood flow follow. Treating diabetic rats with evening primrose oil replenishes PGI2 and PGE2 levels in sciatic nerve by increasing arachidonic acid levels [169] and restores blood flow, preventing nerve hypoxia and normalizing the NCV [246,247]. Vascularization of the endoneurium is augmented by extended treatment with evening primrose oil [248]. Cyclooxygenase inhibitors block this effect, showing the importance of prostaglandins in this process [225]. It seems from these studies that deficiency in fatty acid synthesis exerts its main effect on the endoneural vasculature. The resulting increase in blood flow arising from the correction of essential fatty acid levels is potentiated by subsequent prostanoid synthesis and NO release.
L-Carnitine levels are also reduced in experimental models of diabetes, and replenishment averts the development of diabetic neuropathy [169,249]. L-Carnitine has numerous physiologic effects, but how it specifically prevents diabetic neuropathy is unknown. L-Carnitine assists cellular energy metabolism by facilitating mitochondrial transport of long-chain fatty acids for β-oxidation. Other functions of L-carnitine include restoring Na+, K+-ATPase activity, improving NO synthesis, normalizing prostaglandin levels [169,186], and improving nerve blood flow [178], as well as having possible antioxidant properties [205].
The role of essential fatty acid metabolism and vasoactive prostanoids in the pathogenesis of diabetic neuropathy is important. However, deficits in essential fatty acid probably act together with other metabolic pathways such as the polyol pathway, non-enzymatic glycation, and possibly C-peptide deficiency to compromise vascular and nerve function.
Role of Neurotrophic Factors
Perturbations in the synthesis and function of a variety of neurotrophic factors have been implicated in the development of diabetic neuropathy [250]. Neurotrophins are synthesized by the target cells of particular axons and transported by retrograde transport to the neuronal soma, where they regulate the expression of a variety of proteins. The neurotrophin family includes nerve growth factor (NGF), which is the most widely studied member, but also neurotrophin-3 (NT-3), neurotrophin-4/5 (NT-4/5), and brain-derived neurotrophic factor (BDNF). It appears that each neurotrophic factor may be responsible for maintaining a particular population of neurons within the peripheral nervous system [251]. In the case of NGF, it is synthesized by muscle and skin, then retrogradely transported to DRGs and sympathetic neurons. After transport it can bind one of two receptors, the high-affinity trkA or the low-affinity p75 [251,252]. Diabetic rats experience decreased NGF levels as a result of decreased synthesis in axonal target tissues and decreased axonal transport [253,254]. Furthermore, the expression of both NGF receptors is down-regulated, reflected by decreased trkA levels in DRG and decreased levels of p75 in sciatic nerve [255]. Importantly, these changes do not take place acutely but occur slowly over weeks to months in diabetic rats [250,254].
NGF is responsible for maintaining the production of medium- and low-molecular-weight neurofilaments, and NGF treatment in axotomized DRG up-regulates the mRNA of these structural proteins [256,257]. Insulin has also been shown to up-regulate neurofilament synthesis [258]. The deficiencies in neurotrophic factors are likely to contribute to the characteristic axonal atrophy of diabetic neuropathy, since neurofilaments are the chief determinant of axonal size [259]. Substance P is another protein under the influence of NGF, which may explain the hyperalgesia and allodynia experienced by patients around the injection site of NGF [260,261].
During nerve fiber regeneration the connection of the nerve fiber to its target tissue is lost and NGF support is then provided by Schwann cells and fibroblasts at the site of injury. This response is probably mediated by c-fos and c-jun [250]. NGF elaborated from Schwann cells and fibroblasts is believed to induce interleukin-1 from macrophages that are recruited to the site of injury, and which in turn maintain sustained production of NGF by Schwann cells and fibroblasts [262]. The delayed initiation and the decrease in the sustaining of nerve fiber regeneration in diabetic neuropathy can be attributed to derangement in immediate gene responses, such as the IGF system and c-fos and the markedly decreased local levels of NGF and trkA mRNAs [250]. Other factors leading to inefficient or inadequate nerve fiber regeneration in diabetic neuropathy include decreased induction of growth-associated protein 43 [263]. Far fewer data are available as to the roles of other neurotrophins in diabetic neuropathy. NT-3 may be particularly useful in treating sensory symptoms, and one study showed improvement in sensory NCV in diabetic rats after four weeks of NT-3 treatment [250].
Circulating peptides such as insulin-like growth factor-1 (IGF-I), insulin and C-peptide have neuroprotective effects on peripheral nerve. The majority of IGF-I is synthesized by hepatocytes, but IGF-I is also elaborated by Schwann cells of peripheral nerve, where it has autocrine and paracrine functions. Both hepatic and peripheral nerve IGF-I levels show gradual and progressive declines that can be profound by eight weeks of diabetes in experimental models [172]. In type 1 diabetes, insulin and C-peptide are markedly deficient, whereas in type 2 diabetes normal or even increased serum levels of these hormones occurs. This may explain, at least in part, the tendency for type 1 diabetic neuropathy to be more severe than the neuropathy associated with type 2 diabetes [163]. This concept will be further explored later in this chapter. IGF-I, insulin, and C-peptide all possess neurotrophic actions on sensory, autonomic, and motor neurons [167,264]. It has been suggested that IGF-I synthesis may be compromised by insulin deficiency [264].
The IGF-I receptor (IGF-IR) is expressed on neuronal somata, neurites, and Schwann cells of peripheral nerve, while the insulin receptor (IR) immunolocalizes to paranodal Schwann cells and paranodal axolemma [265]. A C-peptide receptor has not been conclusively identified, but specific binding of C-peptide to cell membranes has been demonstrated by fluorescence correlation spectroscopy [266]. Studies in our laboratory demonstrate that C-peptide possesses insulinomimetic effects and enhances IR phosphorylation and activity [267]. IGF-I has been shown to enhance the synthesis of low- and medium-molecular-weight neurofilaments, and insulin assists in their phosphorylation, which is critical for their polymerization and exportation to the neurites [264]. Insulin, IGF-I, and NGF have been shown to stabilize α- and β-tubulin mRNAs in vivo [258,268]. IGF-I also enhances the motility of neural growth cones by activating focal adhesion kinases and reorganizing actin [269]. ARI-treated type 1 diabetic rats show a six-fold up-regulation of IGF-I mRNA, linking an early metabolic alteration in diabetic neuropathy to the change in neurotrophic factors, which is considered a late change [255].
Neuropathy Associated with Hyperinsulinemia and Hypoglycemia
Some authors suggest that frequent hypoglycemic episodes may play a role in the pathogenesis of diabetic neuropathy [270,271]. Insulinoma patients experience a distal axonopathy that affects both sensory and motor nerves [272]. In transgenic mice with insulinoma a predominantly motor axonopathy ensues [273], while STZ rats harboring an insulinoma develop severe axonal atrophy, degeneration, regeneration, and de-myelination that involves both sensory and motor nerve fibers [274]. Severe hypoglycemia has also been associated with demyelination and anterior horn cell pathology [168,275]. Mohseni et al. examined hypo-and euglycemic type 1 BB/W rats and found mild changes in plantar skin innervation but structurally normal DRGs. In contrast, the ventral roots at L5 showed pathologic alterations, supporting the notion that hypoglycemic neuropathy tends to affect motor neurons [276]. Mohseni et al. also contend that hypoglycemia preferentially affects the nerve trunk, but this is controversial, and the proximal spread of the changes may be variable depending on the severity or duration of hypoglycemic events [277].
It is well documented that strict glycemic control with insulin can precipitate painful neuropathy in diabetic subjects. Painful idiopathic neuropathy is common in patients with impaired glucose tolerance. These observations are consistent with the finding of a small fiber neuropathy in long-term glucose-intolerant GK rats [277a]. In sural nerve biopsies from diabetics with acute painful neuropathy, fluorescein angiography demonstrated epineurial vessel proliferation, increased vascular permeability, and arteriovenous shunting-changes comparable to those of diabetic retinopathy [278]. Vascular endothelial growth factor (VEGF) expression is stimulated in the retina by insulin and is considered an important endothelial cell mitogen, angiogenic factor, and enhancer of vascular permeability. The role of VEGF in diabetic neuropathy has not been tested, but, in rats with insulinoma, insulin levels correlate directly with microvascular density of the endoneurium [279]. These findings tend to suggest a pathogenic role for hyper-insulinemia, particularly in type 2 diabetic neuropathy, which in many respects is distinct from that of type 1 [280,281].
Physiologic Role of Proinsulin C-Peptide
Historically, proinsulin C-peptide was thought to possess no biological function besides that of insulin biosynthesis. However, recent studies compromise the veracity of this claim. The technique of fluorescence correlation microscopy has demonstrated that C-peptide binds to cell membranes in a specific fashion and has an affinity binding constant of 3×l09 M−1 [266]. This means that physiologic concentrations of C-peptide (0.9 nM) saturate the receptor. The nature of the C-peptide receptor remains to be elucidated, but the C-terminal pentapeptide segment of C-peptide appears to be essential for binding and may be the biologically active portion of the molecule [266]. Early reports regarding C-peptide activity showed that an increase in intracellular [Ca2+] occurred followed by activation of Ca2+-dependent protein phosphatase MB, which activates Na+.K+-ATPase by dephosphorylation [282]. Renal tubular cell, sciatic nerve, and erythrocyte Na+, K+-ATPase might be activated in this manner. The rise in intracellular [Ca2+] also activates eNOS [283]. Recent studies in our laboratory show that C-peptide has insulinomimetic effects including up-regulation of glycogen synthesis and amino acid uptake. In rat L6 myoblasts and human neuroblastoma cells (SH-SY5Y), physiologic levels of C-peptide autophosphorylartes the insulin receptor, promoting tyrosine kinase activity. IRS-1 phosphorylation. PI3K activity. MAPK phosphorylation, GSK-3 phosphorylation, and p90Rsk activity [267]. Importantly, an additive effect results from physiologic C-peptide concentrations and submaximal insulin concentrations, but if C-peptide is added in the face of maximal insulin levels it blunts insulin activity [267].
Physiologic roles for C-peptide have been described in a number of tissues. In type 1 BB/W rats, sciatic nerve Na+, K+-ATPase activity was restored by C-peptide replacement [167], which in turn corrected the acute defect in NCV and paranodal swelling that occurs secondarily to a rise in intra-axonal [Na+], the consequences of which will be discussed below (see p 104). Nitric oxide release from bovine aortic endothelial cells is augmented by C-peptide in a concentration-dependent manner [284]. This metabolic effect may underlie the increase in forearm blood flow by C-peptide treatment in type 1 patients and C-peptide-dependent dilation of rat skeletal muscle arterioles [244,285]. The functions of C-peptide are probably intertwined with those of other hormones. The effect of C-peptide on Na+.K+-ATPase activity is potentiated with subthreshold concentrations of neuropeptide Y [282]. Insulin-mediated glycogen synthesis and amino acid uptake are potentiated in vitro within a narrow concentration range by C-peptide (0.1-1.0nM)[267].
Human data regarding the effect of C-peptide are limited, but some studies in type 1 diabetic subjects have been conducted. C-peptide improves erythrocyte deformabilty in type 1 diabetic patients, possibly via enhancing Na+.K+-ATPase function [283]. A 25% increase in glucose utilization is observed in type 1 diabetics under euglycemic clamp when C-peptide levels are normalized [286]. This is congruent with the finding of increased glucose uptake in forearm muscle [287] and muscle strips in vitro from diabetic subjects [288]. Evidence is mounting that C-peptide may be important in the development of some diabetic complications. In type 1 diabetic subjects normalization of C-peptide level is associated with decreased glomerular filtration rate and increased renal plasma flow [286]. When C-peptide was provided continuously by a subcutaneous pump, a decrease in glomerular filtration rate and a marked reduction in urinary albumin resulted [289]. The function of the autonomic nervous system, as assessed by heart rate variability, improved after C-peptide administration [165] and in patients given C-peptide for three months [290]. None of these effects were apparent when insulin was administered alone. No studies have been performed to examine the effect of C-peptide replacement on type 1 diabetic retinopathy.
In the insulin and C-peptide deficient type 1 BB/W rat IR and IGF-IR mRNA are up-regulated in peripheral nerve, but down-regulated in the hippocampus. C-peptide administration to these animals normalizes IR and ICF-1R mRNA levels in peripheral nerve and the central nervous system [265,291]. In addition, hippo-campal apoptosis in the type 1 BB/W rat is prevented by C-peptide, and this may be mediated by up-regulation of hippocampal IGF-IR [291].
How these metabolic effects lead to correction of the structural changes of the peripheral nerve in type 1 diabetic neuropathy is an evolving field of investigation. At this point, we will turn towards describing the structural changes present in experimental diabetic neuropathy and compare the structural changes in type 1 and type 2 animal models.
Relationships Between Metabolic Alterations and Structural Pathology in Type 1 and Type 2 Diabetic Neuropathy
Mounting clinical and experimental evidence indicates that the neuropathy associated with type 1 diabetes is more severe than that associated with type 2 diabetes (Table 4.4) [163,166,292]. Briefly, type 1 diabetic neuropathy is typified by severe axonal atrophy, diffuse fiber loss, axoglial dysjunction, and nodal and paranodal changes. The structural changes of type 2 diabetic neuropathy are characteristically less severe and manifest as mild axonal atrophy, focal fiber loss, segmental demyelination, and Wallerian degeneration. In the past, controversy reigned regarding the nature of diabetic neuropathy, with one camp supporting an underlying axonal disorder and another in favor of a Schwann cell disorder with segmental demyelination as the primary alteration. It now appears that those favoring a primary axonal disorder were describing the changes of type 1 diabetic neuropathy while those supporting a primary Schwann cells disorder were studying the changes of type 2 diabetes [4,15,293].
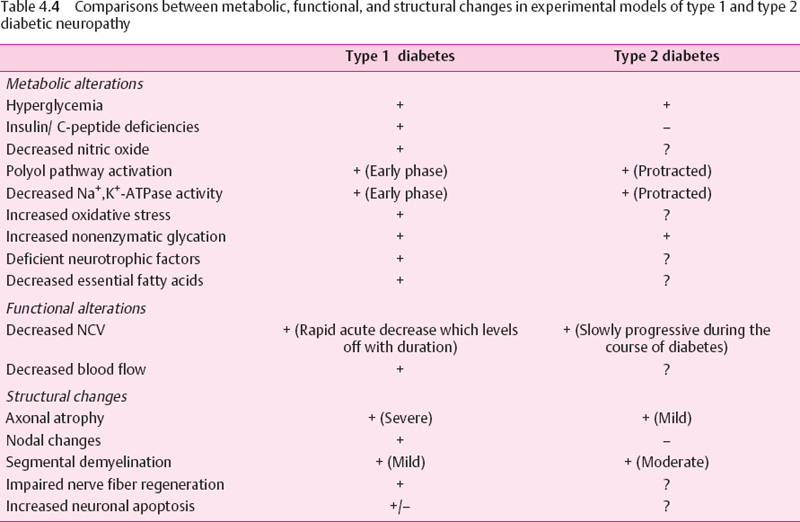

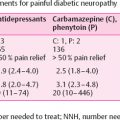

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


