Fig. 18.1
The principle of synthetic lethality applied to cancer medicine. While the loss of either PARP or BRCA function independently does not compromise the ability of the cell to repair DNA damage, PARP inhibition in a cell which has lost BRCA function (right) results in inability to repair the damage and consequently, cellular death
Several PARP inhibitors are at different stages of clinical drug development, basically exploring antitumor activity in patients harboring genetic aberrations in HR-mediated DNA repair. An alternative approach is to take advantage of the capacity of PARP inhibitors to potentiate the effects of DNA-damaging agents, such as certain chemotherapeutics or radiation, by disabling the capacity of the tumor cell to repair the damage induced by platinum drugs or radiotherapy, thereby impacting on the survival of such cells. Several preclinical studies have demonstrated how combination regimens with PARP inhibitors sensitize tumor cells to platinums, anthracyclines, alkylating agents, and topoisomerase inhibitors [13, 14]. PARP is a major effector of DNA strand break repair following damage induced by radiotherapy treatments, which may lead to the development of therapy resistance. There is therefore strong rationale to assess PARP inhibitors as radiosensitizer.
Moreover, aside from its main role in DNA repair there is evidence that PARP-1 is involved in the transcription regulation of the androgen receptor (AR) and in the rearrangement of specific genes, such as ERG, ETV1, and FLI1 [16–18]. Studies demonstrating robust preclinical antitumor activity in models expressing TMPRSS2-ERG rearrangements, together with evidence of the suppression of AR-targeted gene expression, support the targeting of PARP in prostate cancer [15–17].
The Target Population for PARP Inhibitors
Although loss of a variety of genes involved in HR DNA repair may sensitize to PARP inhibition, to date the only predictive biomarker of response to PARP inhibitors that has been validated in clinical trials is the presence of germline mutations in the BRCA1/2 genes.
BRCA1 and BRCA2 are both tumor suppressor genes coordinating mechanisms of response to DNA damage through HR-mediated DNA repair. While BRCA2 function seems to be limited to DNA repair control, generally through the regulation of RAD51, BRCA1 may have an additional range of functions, with effects over cellular control systems, chromatin modeling, and the regulation of several transcription factors [18–20].
BRCA1/2 (germline) mutation carriers with prostate cancer currently undergo treatment similar to patients with sporadic prostate cancer. This is despite the fact that prostate cancer in BRCA mutation carriers has a more aggressive phenotype. They usually present with a higher Gleason score, develop nodal and metastatic involvement more frequently, and have worst survival outcomes [21, 22]. Nevertheless, their rates of response to taxane-based chemotherapy are not dissimilar to the general population [23]. The question as to whether such patients harbor a genetically defined subset of prostate cancer that is responsive to platinum-based chemotherapy remains to be elucidated.
BRCA2 mutations confer an 8.6-fold increase in the risk of developing prostate cancer in men up to 65 years of age, while BRCA1 mutations increase the risk by 3.5-fold [24, 25]. In animal models, BRCA2 dysfunction has been shown to induce the appearance of premalignant prostate lesions, but other genetic events are believed to be necessary for carcinogenesis [26]. For example, it is hypothesized that genomic instability secondary to DNA repair impairment paves the way for other oncogenic events, including those present in prostate cancer.
Interestingly, since the BRCA protein acts also as a regulator of the AR pathway, there is increasing interest in combining DNA repair-targeting agents with AR-axis directed therapies. Similarly, crosstalk between the phosphatidylinositol 3-kinase (PI3K)–AKT pathway and BRCA1 provides a strong rationale for the investigation of combination regimens of PARP inhibitors and drugs targeting the PI3K–AKT signaling network [27–29].
Based on strong preclinical rationale demonstrating tumor-specific antitumor activity of PARP inhibitors in BRCA1/2 mutant cells [9, 10], a phase I proof-of-concept study of olaparib (KU-0059436; AZD2281; AstraZeneca) was commenced. The study population was initially enriched with germline BRCA mutation carriers, but following the observation of objective antitumor responses in this subgroup of patients, recruitment in the phase I dose expansion cohorts was limited to this genetically defined population [30].
Since then, a number of clinical trials have tested the antitumor efficacy of olaparib and other PARP inhibitors both in germline BRCA1/2 mutation carriers and sporadic cancer populations [31–35]; Gelmon and colleagues reported a clinical trial on olaparib for patients with advanced ovarian or triple negative breast cancers, stratifying them on the basis of the presence of germline BRCA1/2 mutations [36]. Among sporadic (BRCA1/2 wildtype) patients, there were no objective responses in patients with breast cancer. However, patient benefit was observed in those with platinum-sensitive ovarian cancer, due to the induction of DNA damage. The expression of certain DNA repair markers, which may be impaired in BRCA1/2 mutant cells, is associated with increased sensitivity to platinum-based chemotherapy [37].
Germline mutations in BRCA1/2 genes are inherited in an autosomal dominant manner, with incomplete penetrance, and are present in less than 2 % of sporadic prostate cancers [25, 38]. Therefore, the target population that is likely to have greatest potential to benefit from PARP inhibitors may be small, in comparison with triple negative breast cancer where the prevalence of BRCA1/2 mutations ranges between 10 and 20 % in unselected populations [39, 40]. There is therefore great interest in finding other potential response biomarkers of response to PARP inhibition through studies investigating the DNA repair system. Evidence of clinical activity in patients who are not BRCA1/2 mutation carriers, such as sporadic high grade ovarian cancer and sporadic CRPC supports a wider role for PARP inhibitors [36, 41]. Theoretically, the application of the concept of synthetic lethality to PARP inhibition would not only be limited to BRCA1/2 germline mutation carriers, but also include patients with functional loss of DNA repair capacity through alternative mechanisms [42]. Importantly, BRCA1/2 mutations only account for a fraction of known defects in HR-mediated DNA repair. Sensitivity to PARP inhibitors in preclinical models with genetic and epigenetic aberrations involved in the response to DNA damage has led to an armamentarium of potential markers that now need to be evaluated in the clinical setting including: (a) the phosphatase and tensin homolog (PTEN) gene not only through its role as a negative regulator of PI3K but as a warrant of chromosomal integrity and regulator of RAD51 transcription [43]; (b) tumors with other DNA repair genomic aberrations including deficiencies in ATM, ATR, CHEK2, RAD51, PALB2, and other FANC genes [12, 44]; and (c) epigenetic alterations silencing wild-type BRCA1/2 and other DNA repair genes [45]. The critical interaction between PARP1, DNA-PKc and the resulting protein of gene fusions, especially of the androgen-responsive gene transmembrane protease serine 2 (TMPRSS2) with the oncogenic erythroblast transformation specific (ETS) transcription factor family of genes, present in approximately 50 % of prostate cancers, has generated great interest in evaluating this recurrent fusion protein in prostate as a potential biomarker of sensitivity to DNA repair targeting [16].
In order to optimize the application of PARP inhibitors, there is an urgent need to develop and validate clinical biomarkers that interrogate the functionality of HR-mediated DNA repair systems. Such assays may use gene expression and/or protein transcription profiles to identify tumors not known to harbor BRCA1/2 mutations, but which express similar biological features, a phenotype coined as a “BRCAness” profile. A potential approach is the use of large short-interfering RNA (siRNA) panels to screen potential predictors of PARP inhibitor sensitivity [46].
It is critical that clinical trials evaluating PARP inhibitors in prostate cancer implement functional assays so as to characterize their pharmacodynamic effects and aid in the identification of predictive biomarkers of PARP blockade. Such strategies may evaluate the formation of RAD51 and γH2AX foci in tumor and surrogate tissue, including circulating tumor cells, peripheral blood mononuclear cells, and hair follicles.
Clinical Experience with PARP Inhibitors
The proof-of-concept phase I study of olaparib prospectively enriched each dose escalation with patients harboring germline BRCA1/2 mutations before restricting accrual to patients with BRCA1/2 mutant tumors in the dose expansion phase [30]. The study identified fatigue, mood alteration, somnolence, and thrombocytopenia as dose-limiting toxicities. There were no significant differences in the toxicity profile between BRCA1/2 mutation carriers compared to WT BRCA1/2 patients. Establishment of the biologically active dose-range of olaparib was guided by parallel pharmacokinetic and pharmacodynamic evaluation of normal tissue, including peripheral blood mononuclear cells from blood, hair follicles, and tumor tissue samples. The dose selected from this dose-escalation study was 400 mg twice daily and although 100 mg was demonstrated to have biologically relevant effects, subsequent studies have shown a dose–response relationship for olaparib efficacy. The higher dose of 400 mg BD has therefore been utilized as the preferred dose for later-stage studies [33, 35].
Significant evidence of antitumor activity was observed in this phase I trial among BRCA1/2 mutations carriers, as predicted from preclinical data [9]. There was evidence of antitumor activity in germline BRCA1/2 mutation carriers suffering from CRPC, including a patient who had a response lasting for almost 3 years, including complete radiological resolution of bony metastases and continued prostate specific antigen (PSA) tumor marker response (Fig. 18.2).
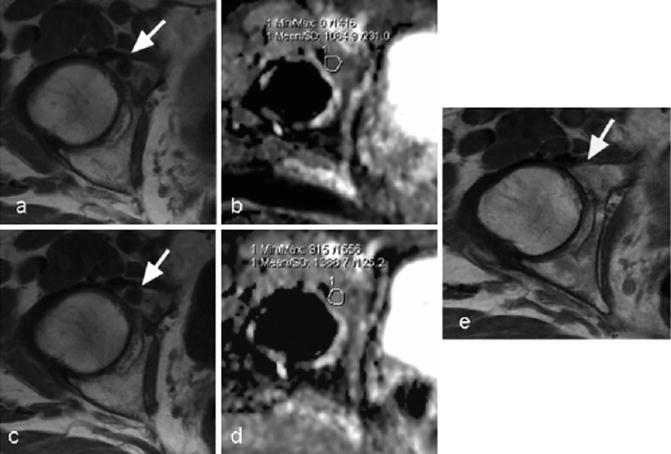
Fig. 18.2
Diffusion-weighted magnetic resonance imaging (MRI) demonstrating disease regression in a patient with prostate cancer and BRCA2 mutation associated with a >50 % decline in PSA. A 63-year-old man with germline BRCA2 mutation and castration-resistant prostate cancer. T1 weighted MRI at the level of the right acetabulum in the pelvis obtained (a) prior to and (c) 3 months after initiating treatment with olaparib showed no substantial change in a 13 mm low-signal intensity (dark) metastasis in the right acetabulum (arrows). Apparent diffusion coefficient values increased from pretreatment (b) to after 3 months of treatment (d) >30 % at the site of metastatic disease (circled) consistent with disease regression. (e) T1-weighted imaged 1 year after starting treatment showing resolution of disease (arrow). Images courtesy of Dr. Dow-Mu Koh, The Royal Marsden NHS Foundation Trust, UK. Adapted from Fong et al., NEJM 2009
Several phase II studies of olaparib have now been pursued; overall, the compound has been shown to induce substantial antitumor activity as a single agent in BRCA1/2 mutation carriers with ovarian and breast cancer, as well as several cases of sporadic ovarian cancer. In this later tumor type, analysis of a series of BRCA1/2 mutation carriers demonstrated cross-sensitivity between prior platinum therapies and PARP inhibitors [34]. Conversely, patients who develop disease progression on PARP inhibitors have been found to still have the potential to respond to further lines of platinum-based chemotherapy [47].
Pivotal studies evaluating the role of olaparib at different stages of ovarian cancer and other tumor types are ongoing or planned, either as monotherapy or in combination with other antitumor agents. There is also an ongoing two-stage phase II study evaluating the antitumor activity of olaparib in unselected patients with advanced CRPC. This study aims to identify predictive biomarkers to guide appropriate patient selection that would be validated in a subsequent cohort of patients (NCT01682772) (Fig. 18.3).
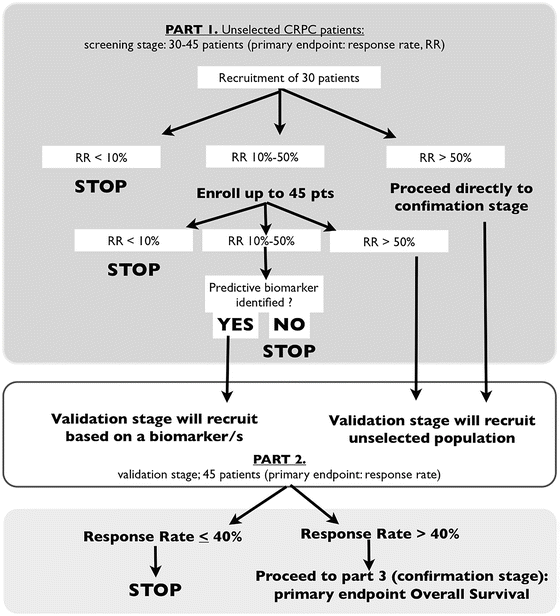
Fig. 18.3
Adaptative design of the TOPARP study, an open-label phase II study of olaparib in CRPC patients to evaluated antitumor activity and investigate predictive biomarkers
Veliparib, previously known as ABT-888 (Abott Laboratories), was shown to inhibit PARP-1 and PARP-2 in preclinical models and to potentiate the effects of DNA-damaging agents, such as cytotoxics or radiation [13, 48]. An initial phase 0 trial was conducted, comprising the administration of single doses of veliparib to patients with advanced cancers, followed by the assessment of pharmacokinetics and pharmacodynamics through subsequent tumor biopsies and surrogate tissue analysis [49]. This aided in the design of dose-escalation trials of veliparib in combination with different chemotherapies. Among the numerous combinatory trials in different tumor types, a phase II study is assessing the combination of veliparib with abiraterone acetate in patients with CRPC (NCT01576172). A separate study treated 25 CRPC patients with veliparib and temozolomide; of these patients, one had a 37 % decrease in PSA and another had a 97 % decrease in PSA associated with a radiological response [50].
The phase I trial of niraparib (MK-4827; Merck, Tesaro) enrolled 18 patients with sporadic CRPC in an expansion cohort at the maximum tolerated dose (300 mg QD) and three further treated during dose escalation. This study recruited a total of 60 patients with advanced solid tumors in the dose-escalation stage, including 29 patients with BRCA1/2 mutations; the latter group of patients included 22 with advanced ovarian or primary peritoneal cancer, four patients suffering from breast cancer and individual cases of pancreatic, lung, and prostate cancers. A second stage of the study recruited a further 22 patients with sporadic ovarian carcinoma and the aforementioned 18 sporadic CRPC patients, on the basis of the relevance of DNA repair defects in the pathogenesis of these diseases [41].
Pharmacodynamic studies showed that PARP inhibition in peripheral blood mononuclear cells exceeded 50 % at most doses greater than 80 mg/day. Induction of γH2AX foci in circulating tumor cells and a limited number of paired tumor biopsies was demonstrated. 8/20 (40 %) and 2/4 (50 %) BRCA1/2 mutation carriers with ovarian and breast cancer, respectively, achieved radiological partial responses. Antitumor radiological and/or biochemical responses were detected in 5 of the 22 patients with sporadic ovarian cancer, predominantly in those with platinum-sensitive disease.
Among the 21 patients with CRPC, nine had radiological stability for more than 4 months, with a median duration of treatment of 254 days in this subgroup. One patient experienced >50 % decrease in PSA on treatment, remaining on treatment for 10 months. Three patients had significant declines in CTC counts, which were maintained for at least 8 months prior to disease progression. Interestingly, one of the three patients participating in the dose-escalation phase of the study was a BRCA1/2 mutation carrier who did not benefit from niraparib. This study included several biomarker-finding studies for PARP inhibitors in prostate cancer, including the assessment of PTEN function and the presence of ERG fusions in archival tumor samples and circulating tumor cells from 18 CRPC patients in the study. No correlation between PTEN and ERG status with decreases of PSA or time to tumor progression was found.
Rucaparib (AG-014699/CO-338, Pfizer/Clovis Oncology) is another oral PARP1/2 inhibitor that has been assessed in two dose-escalation studies; one as monotherapy and another in combination with several chemotherapy regimens in a population enriched with but not limited to BRCA1/2 mutation carriers [51, 52]. In preclinical studies, rucaparib induced selective cytotoxicity in tumor cells that were defective in HR-mediated DNA repair [53]. The recruitment of patients with sporadic cancers is supported by preclinical studies demonstrating enhancement of chemotherapy effects in different ovarian cancer cell lines with alternative gene aberrations related with DNA repair mechanisms, such as loss of PTEN function, low expression of RAD51, or silencing by methylation of wildtype BRCA [54]. An intravenous (IV) formulation had previously been evaluated in a dose escalation study in patients with BRCA1/2 mutant ovarian and breast cancers. Preliminary clinical studies reported an overall response rate of 5 % at dose levels evaluated, which are lower than equivalent oral doses investigated. The IV formulation was also evaluated in combination with chemotherapy, such as with temozolamide in a clinical trial in patients with advanced melanoma [55].
Preliminary results of a first-in-human study on the PARP inhibitor BMN-673 (BioMarin) were reported at the 2013 ASCO Annual Meeting [56]. BMN-673 demonstrated high potency in inhibiting PARP in preclinical studies and showed antitumor cytotoxicity in cells with deficient BRCA1/2 or PTEN function [57]. Overall, initial reports of the trial showed good oral bioavailability for BMN-673, which induced tumor responses in patients with germline BRCA1/2 mutant breast and ovarian cancers. Hematological events were dose-limiting, resulting in the selection of 1,000 mcg QD as the dose for further development, which was tenfold above the minimum dose to show target modulation.
As PARP inhibitors are implemented in clinical practice, our understanding of the underlying mechanisms of both primary and secondary resistance will be even more relevant and critical for the optimal application of this class of drugs. A common feature to all molecular targeted agents developed over the past decade is the inevitable development of drug resistance, mostly through tumor evolution in the context of selection pressures induced by prolonged drug exposure. For PARP inhibitors, mechanisms of resistance may include expulsive pumps that decrease intracellular drug availability, a progressive reliance of the cells on alternative mechanisms of DNA repair, and restoration of BRCA1/2 function through gene reversion [58, 59].
Related posts:
 Molecular Mechanisms of Prostate Cancer Progression After Castration
Targeting C-Met/VEGF in Castration Resistant Prostate Cancer
Molecular Mechanisms of Prostate Cancer Progression After Castration
Targeting C-Met/VEGF in Castration Resistant Prostate Cancer
 Cytotoxic Chemotherapy (Taxanes and Taxane Combinations)
Cytotoxic Chemotherapy (Taxanes and Taxane Combinations)
 Phase I–II Targeted Treatments
Phase I–II Targeted Treatments
 Epigenetics in Castration Resistant Prostate Cancer
Epigenetics in Castration Resistant Prostate Cancer
 Radium-223 and Other Radiopharmaceuticals in Prostate Cancer
Radium-223 and Other Radiopharmaceuticals in Prostate Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


