Paroxysmal nocturnal hemoglobinuria is manifests with a chronic hemolytic anemia from uncontrolled complement activation, a propensity for thrombosis and marrow failure. The hemolysis is largely mediated by the alternative pathway of complement. Clinical manifestations result from the lack of specific cell surface proteins, CD55 and CD59, on PNH cells. Complement inhibition by eculizumab leads to dramatic clinical improvement. While this therapeutic approach is effective, there is residual complement activity resulting from specific clinical scenarios as well as from upstream complement components that can account for suboptimal responses in some patients. Complement inhibition strategies are an area of active research.
Key points
- •
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, clonal, hematopoietic stem cell disorder with 3 clinical features: hemolytic anemia from uncontrolled complement activation, thrombosis, and bone marrow failure.
- •
Eculizumab is a humanized monoclonal antibody that binds to C5 in complement system and decreases intravascular hemolysis, reduces thrombosis risk, and improves quality of life.
- •
Persistent extravascular hemolysis in PNH while on eculizumab remains a relevant clinical issue and multiple therapies are being examined to improve this.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, clonal, hematopoietic stem cell disorder that manifests with a hemolytic anemia from uncontrolled complement activation, bone marrow failure, and a propensity for thrombosis. It is the chronic hemolytic anemia in PNH, largely mediated by the alternative pathway of complement (AP), from which the disease derives its descriptive moniker. PNH is a unique disease whose clinical manifestations have been linked to the deficiency in glycosylphosphatidylinositol-anchored proteins (GPI-APs). These manifestations include a lack of the complement regulatory proteins CD55 and CD59. CD55 regulates the formation and stability of the C3 and C5 convertases, whereas CD59 blocks the formation of the membrane attack complex (MAC).
The bone marrow failure component of the disease is well-appreciated. The mechanism of the thrombophilia is less well-described. Historically, PNH is among the first diseases in which the role the complement cascade plays in the pathogenesis is well-elucidated. This review focuses on the dysregulation of the complement cascade, leading to the hemolytic anemia in PNH as well as its other clinical manifestations and the therapies available presently and possibly in the future for the disease.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, clonal, hematopoietic stem cell disorder that manifests with a hemolytic anemia from uncontrolled complement activation, bone marrow failure, and a propensity for thrombosis. It is the chronic hemolytic anemia in PNH, largely mediated by the alternative pathway of complement (AP), from which the disease derives its descriptive moniker. PNH is a unique disease whose clinical manifestations have been linked to the deficiency in glycosylphosphatidylinositol-anchored proteins (GPI-APs). These manifestations include a lack of the complement regulatory proteins CD55 and CD59. CD55 regulates the formation and stability of the C3 and C5 convertases, whereas CD59 blocks the formation of the membrane attack complex (MAC).
The bone marrow failure component of the disease is well-appreciated. The mechanism of the thrombophilia is less well-described. Historically, PNH is among the first diseases in which the role the complement cascade plays in the pathogenesis is well-elucidated. This review focuses on the dysregulation of the complement cascade, leading to the hemolytic anemia in PNH as well as its other clinical manifestations and the therapies available presently and possibly in the future for the disease.
The pathophysiology of the complement dysregulation in paroxysmal nocturnal hemoglobinuria
The complement system is our host defense system that protects the intravascular space through opsonizing and lysing bacteria. The complement system consists of plasma proteins that interact via 3 major pathways: the classical, alternative, and lectin binding. This system encompasses these distinct cascades with individual functions, which all converge into a common final effector mechanism—the MAC ( Fig. 1 ). These 3 pathways independently lead to activation of C3 and C5 convertases. Although the classical and the lectin pathways require specific triggers to be activated—usually infection—it has been known for years that the AP exhibits low-grade continuous activation owing to spontaneous hydrolysis of C3 (called the “tick-over” phenomenon). In addition, some components of the AP constitute an amplification mechanism (the so-called AP amplification loop), which amplifies complement activation regardless of the specific pathway that initially generates the first C3b molecule (see Fig. 1 ). Multiple mechanisms have evolved to control the complement cascade, including membrane-bound proteins (complement receptor 1 [CR1], membrane cofactor protein, and the membrane proteins CD55 and CD59), as well as fluid-phase components, including complement factor I and factor H (FH). Among these, CD55 and CD59 are of pivotal importance in PNH, given that they are normally expressed on hematopoietic cells and attached by the GPI anchor proteins.
With specific respect to the thrombosis seen in PNH, there are many direct and bidirectional interactions between the complement system and the coagulation cascade (see Fig. 1 , which shows where thrombin interacts). Most notable to the clinical implications is that thrombin can cleave C5 into C5a, which occurs independent of C3 and therefore represents a bypass of the 3 traditional complement activation pathways (that is, the classical, lectin, and APs). Thrombin-activatable fibrinolysis inhibitor inactivates C3a and C5a in a negative feedback loop. The complement system also amplifies coagulation through the C5a-mediated induction of expression of tissue factor and plasminogen activator inhibitor 1 by leukocytes, the latter of which inhibits fibrinolysis. This activation of the complement through the generation of thrombin contributes to hypercoagulability in PNH and may explain why thrombosis in PNH often leads to an inexorable disease flare that is best interrupted by blocking terminal complement.
The complement proteins in paroxysmal nocturnal hemoglobinuria
PNH erythrocytes are highly vulnerable to complement-mediated lysis owing to a reduction, or absence, of 2 important GPI-anchored complement regulatory membrane proteins (CD55 and CD59). CD59 is a glycoprotein that directly prevents the MAC from perforating the cell membrane by blocking the aggregation of C9. CD55 accelerates the rate of destruction of membrane-bound C3 convertase. CD55 (also called decay accelerating factor) inhibits the formation and the stability of C3 convertase (both C3bBb and C4b2a). CD55 was the first complement regulator reported to be absent on PNH erythrocytes. CD59 was later identified as an additional complement regulatory protein on PNH erythrocytes. Distinct from CD55, CD59 interferes with the terminal effector complement, blocking the incorporation of C9 onto the C5b–C8 complex, forming the MAC. CD59 is the more important molecule of the two—because, if it is absent, this leads to lysis, whereas an isolated deficiency of CD55 can be overcome. There are patients with a congenital isolated CD55 deficiency but normal CD59 expression who do not hemolyze (see Fig. 1 ).
Paroxysmal nocturnal hemoglobinuria as a hemolytic anemia
PNH erythrocytes’ in vitro susceptibility to hemolysis was initially described by Dr Ham in his pivotal studies in the 1930s. Dr Ham demonstrated that erythrocytes from patients who have PNH hemolyze in their own serum, especially when the AP is activated (by acidification). This hemolytic assay is called the Ham test or the acidified serum assay. This feature of PNH erythrocytes was subsequently described further when it was demonstrated that a distinct phenotype of PNH erythrocytes may exist, according to their specific sensitivity to complement-mediated lysis in vitro. In fact, in PNH there are erythrocytes with only modest hypersensitivity (3–5 times normal values) or a more pronounced hypersensitivity to complement-mediated lysis (15–25 times the normal one). These phenotypes are now known as PNH type II and type III erythrocytes, which by flow cytometry correspond with a complete (type III) or partial (type II) deficiency of GPI-APs, respectively. Chronic hemolysis of PNH is likely owing to a continuous steady-state complement activation coming from the low-grade spontaneous C3 tick-over, with ongoing activation of the CAP, as described. Infections or inflammatory status usually result in hemolytic crises (the paroxysms that given the disease its name), eventually as a result of substantial complement activation.
Paroxysmal nocturnal hemoglobinuria as a disease of marrow failure
Patient with PNH obviously suffer from anemia, but often they also have other cytopenias in the setting of their marrow failure owing to impaired hematopoiesis. The marrow failure component of PNH can vary from subclinical disease to severe aplastic anemia (AA) and may be categorized as such. It has been demonstrated that PNH patients have a reduced number of hematopoietic progenitors assessed by cultures assays regardless of the categorization. There is considerable overlap between PNH and AA both in disease presentation; in addition, they have long been viewed as distinct presentations of the same disorder. The mechanism by which PNH clones expand has been an area of active research. Immunoselection is considered to be essential for the selective expansion of these clones. The expansion is not simply attributable to PIGA mutations alone. Also, PNH clonal populations can be detected frequently in patients with the other marrow failure syndromes, such as AA and myelodysplastic syndrome. This may suggest that GPI − cells survive immune-mediated bone marrow injury putatively caused by cytotoxic cells such as natural killer cells. Human leukemic K562 cells become relatively resistant to natural killer cell-mediated cytotoxicity after acquisition of PIGA mutations in vitro. This relative survival advantage may be owing to deficiency of stress-inducible GPI-linked membrane proteins upregulation of UL16-binding protein (ULBP)1 and ULBP2 on PNH cells. ULBPs activate natural killer and T cells and are detected on GPI-expressing but not on GPI-deficient K562 cells. Thus, in the setting of an immune attack on the bone marrow, the lack of ULBPs may contribute to immunoselection of the PNH clone over normal cells. There also is the view that the patients with PNH clones and the autoimmune phenomenon of AA have an attack against the hematopoiesis at the level of the stem cell, and this allows the clonal expansion for the clinical PNH phenotype. More recently, it has been suggested that the GPI-AP could be the target of the immune attack and thus the PNH cells are spared naturally, again allowing their clonal outgrowth over the normal hematopoiesis. The PNH clone is often considered a marker of an immune form of marrow failure because it may predict response to immunosuppressive therapy in AA and patients with inherited forms of AA lack the PNH clone. The size of the PNH clone may vary over time and this is the best determinant of the hemolytic component of the disease. Therapies directed at this hemolysis will not improve the patients’ marrow failure.
Paroxysmal nocturnal hemoglobinuria as a disease of thrombosis
Thrombosis is another typical manifestation of PNH. It is the leading cause of death in the disease. Thrombosis may occur at any site in PNH: venous or arterial. Common sites include intraabdominal (hepatic, portal, splenic or mesenteric) and cerebral (cavernous or sagittal sinus) veins, with hepatic vein thrombosis (also known as Budd–Chiari syndrome) being the most common. Deep venous thrombosis, pulmonary emboli, and dermal thrombosis are also prevalent. In contrast with the mechanisms of the hemolysis or the marrow failure, less is definitively known about the pathophysiology and mechanism of the thrombophilia in PNH, especially in patients not treated with eculizumab. Clinically, the complication of thrombosis is more prevalent in patients as the PNH clone increases in size. Thrombosis may occur in any PNH patient, but those with a large percentage of PNH cells (>50% granulocytes) are at greatest risk. This may suggest that the ultimate etiology of the thrombophilia in PNH is related to the hemolysis with complement activation. As discussed, there are also clear interactions between the complement system and the coagulation cascade, namely thrombin and C3, which contribute to the thrombosis in PNH. There are currently several hypothesized mechanisms and ultimately the pathophysiology may be multifactorial. The thrombophilia may directly result from the hemolytic anemia as the free hemoglobin is released by the erythrocytes into the serum causing nitric oxide (NO) scavenging and thus preventing the inhibition by NO on platelet aggregation and adhesion to endothelium. Next, the uncontrolled complement regulation on platelet surface could be hypothesized to lead to platelet activation and aggregation, enhancing the formation of thrombi. Another known issue is that the absence of GPI-APs on PNH platelets leads to thrombotic microparticles. Another possible mechanism of thrombosis in PNH could be a disruption of the fibrinolytic system, owing to the lack of membrane-bound urokinase-type plasminogen activator receptor, another GPI-anchored protein, leaving excess of its soluble form. Complement activation also contributes to the prothrombotic tendency of PNH patients. Specifically, C5a may result in proinflammatory and prothrombotic processes by generating inflammatory cytokines such as interleukin-6, interleukin-8, and tumor necrosis factor. It is unclear which of these mechanisms contribute most to thrombosis in PNH; however, complement inhibition with eculizumab is the most effective means to stop thrombosis in PNH. Anticoagulation and eculizumab are indication for acute thrombotic events; however, primary prophylactic anticoagulation has not been proven to be beneficial in PNH. Anticoagulation after the acute event in a PNH patient well-maintained on eculizumab may not be necessary.
Paroxysmal nocturnal hemoglobinuria and consequences of nitric oxide
Many manifestations of PNH result from intravascular hemolysis and are explained by hemoglobin-mediated NO scavenging after free hemoglobin is released from hemolyzed cells. NO is a major regulator of vascular physiology. NO acts on the vascular wall to maintain normal tone and limit platelet activation. Free hemoglobin has enormous affinity for NO and can reduce the plasma level of NO to the point of causing symptoms. This reduction has been demonstrated in clinical trials where the administration of cell-free hemoglobin solutions to healthy people is associated with development of abdominal pain and esophageal spasm. Under normal conditions, hemoglobin is sequestered by the erythrocyte membrane, which minimizes the scavenging of NO. In PNH, the intravascular hemolysis results in release of large amounts of free hemoglobin into the plasma. This release leads to scavenging of NO and degradation of the substrate for NO synthesis. This depletion of NO at the tissue level manifests clinically as fatigue, abdominal pain, esophageal spasm, erectile dysfunction, and possibly thrombosis. These clinical symptoms are more common in patients with PNH who have larger populations of PNH cells (>60% of granulocytes). Additionally, chronic kidney disease and pulmonary hypertension are complications that may go unrecognized, but also result from scavenging of NO. For example, in pulmonary arterial hypertension the symptoms are usually mild and are often nonspecific (eg, tiredness, breathlessness). Chronic kidney disease stages 1 through 3 are also described and can be quite common in PNH patients.
Diagnosis and classification of paroxysmal nocturnal hemoglobinuria
The diagnosis of PNH is both a laboratory and a clinical diagnosis. The laboratory measures include a reticulocyte count, lactate dehydrogenase (LDH) levels, complete blood count indicative of hemolysis, and peripheral blood flow cytometry to detect the deficiency of the GPI ( Box 1 ). This absence of GPI-APs is detected after staining cells with monoclonal antibodies and a reagent known as fluorescein-tagged proaerolysin variant that binds the glycan portion of the GPI anchor. The erythrocytes may be classified as types I, II, or III PNH cells, as noted. Type I cells have normal levels of CD55 and CD59, whereas type II have reduced levels and type III have complete absence. Hematopathologists have recently published guidelines for diagnosis of PNH using flow cytometry.
Diagnosis
- •
PNH by FLAER assay
- •
LDH
- •
Reticulocyte count
- •
CBC
Therapy
- •
Eculizumab intravenously
- ○
Loading: 600 mg weekly × 4 weeks
- ○
Maintenance (followed 1 week later): 900 mg every 2 weeks thereafter
- ○
- •
Consideration of HSCT in suboptimal responders
Monitoring while on therapy
- •
At least monthly
- ○
LDH, reticulocyte count, CBC, chemistries
- ○
- •
At least yearly
- ○
PNH by FLAER assay
- ○
- •
If concern for extravascular hemolysis
- ○
Direct antiglobulin test
- ○
Abbreviations: CBC, complete blood count; FLAER, fluorescein-tagged proaerolysin variant; HSCT, hematopoietic stem cell transplantation; LDH, lactate dehydrogenase; PNH, paroxysmal nocturnal hemoglobinuria.
Laboratory testing for diagnosis and monitoring during treatment. Standard therapy regimen with eculizumab.
The classification of PNH has been proposed by the International PNH Interest Group (IPIG) and includes 3 subtypes: classical PNH, which includes hemolytic and thrombotic patients who have evidence of PNH in the absence of another bone marrow failure disorder; PNH in the context of other primary bone marrow disorders, such as AA or myelodysplastic syndrome; and subclinical PNH, in which patients have small PNH clones but no clinical or laboratory evidence of hemolysis or thrombosis.
Current therapies for paroxysmal nocturnal hemoglobinuria directed against complement
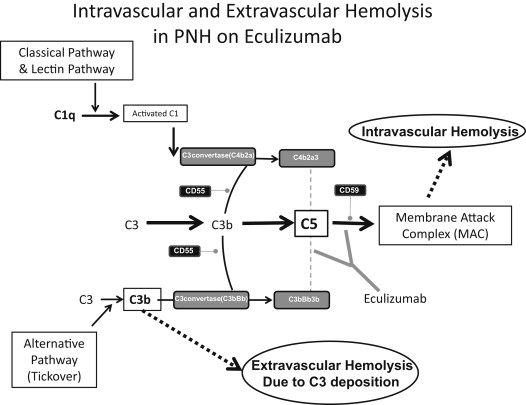
Eculizumab is a humanized monoclonal antibody that binds to C5 and inhibits its further cleavage into C5a and C5b. The drug decreases intravascular hemolysis, reduces thrombosis risk, and improves quality of life in PNH by inhibiting the formation of the MAC ( Fig. 2 ). It is the only therapy approved by the US Food and Drug Administration for PNH.
Related posts:
 Complement in Health and Disease
Complement in Health and Disease
 Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
 Congenital CD59 Deficiency
Warm Autoimmune Hemolytic Anemia
Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia
Current and Future Pharmacologic Complement Inhibitors
Congenital CD59 Deficiency
Warm Autoimmune Hemolytic Anemia
Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia
Current and Future Pharmacologic Complement Inhibitors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


