Fig. 6.1
The location and frequency of ectopic superior (a) and inferior (b) parathyroid glands. The shaded area indicates the embryologic of descent of the parathyroid glands
Adenocarcinoma of the parathyroid is defined by gross or histologic invasion of blood vessels, perineural tissue, thyroid gland, or other surrounding tissues and by the presence of distant metastases. Of note, fibrosis or mitotic figures can be found in adenomas without malignant potential, so these findings are not sufficient to diagnose parathyroid adenocarcinoma [3].
Clinical Presentation
History and Symptoms
Patients with hyperparathyroidism can have a wide range of symptoms, or they can be asymptomatic (Table 6.1) [1, 12]. Pediatric patients are more likely to present with symptoms than adults. While most adult series demonstrate asymptomatic rates of 30–40%, pediatric series report only 0–20% of patients as asymptomatic on presentation [1, 2, 12]. Children are also more likely to present with end organ damage, including pathologic bone fractures, osteitis fibrosa cystica, nephrolithiasis, and pancreatitis [12]. These presentations of advanced disease may be due in part to the frequent delays in diagnosis caused by a low index of suspicion and delay in appropriate confirmatory diagnostic investigations. Hyperparathyroidism is the third most frequent endocrine disease in adults (after diabetes and thyroid disease) [3] and is a commonly considered diagnosis, but in a large series of children and adolescents it took an average of 24 months to diagnose the same disease in pediatric patients [1].
Table 6.1
Clinical presentations of hyperparathyroidism
Asymptomatic |
|---|
General |
• Fatigue |
• Weakness |
• Myalgias |
Neurologic/Psychiatric |
• Headache |
• Depression |
• Cognitive impairment |
Skeletal |
• Bone pain |
• Osteoporosis |
• Pathologic fractures |
• Osteitis fibrosa cystica |
Gastrointestinal |
• Anorexia |
• Nausea |
• Vomiting |
• Diarrhea |
• Constipation |
• Pancreatitis |
• Peptic ulcer disease |
Renal |
• Polyuria |
• Polydipsia |
• Kidney stones |
• Hypertension |
In the rare patients with neonatal severe hyperparathyroidism, symptoms present in the first few days of life and include failure to thrive, hypotonia, respiratory distress, and prominent skeletal involvement. This is always combined with severe hypercalcemia that often requires urgent intervention [2].
A family history of parathyroid disease is common in pediatric patients with HPT, especially in those with genetic disorders such as MEN1 or MEN2, or familial HPT. Patients with these inherited conditions are even more likely to present with symptoms when compared to those without a family history [1, 3, 11, 12].
Physical Examination
The physical examination in children and adolescents with HPT is usually normal. The parathyroid glands lie posterior and medial to the lateral border of the thyroid gland (see Chap. 5) and are difficult to palpate even when enlarged. When patients with HPT have palpable neck nodules, the nodules are usually not parathyroid glands [1]. Occasionally an ectopic gland may be palpated as a small (≤1 cm), mobile, non-tender neck nodule. Cervical lymph nodes are rarely enlarged and supraclavicular lymph nodes are even more rarely enlarged.
Blood Tests
In any child with suspected hypercalcemia and/or hyperparathyroidism, serum levels of total calcium, PTH, phosphate, and alkaline phosphatase should be checked. Serum TSH levels should be checked to rule out concomitant thyroid disease. Diagnosis of primary HPT is made with elevated PTH levels (>65 pg/mL, though the normal range varies by testing protocol) in the presence of normal or elevated serum calcium (>10.2 mg/dL, though the normal range varies by testing protocol). A PTH level that is not appropriately suppressed with a high-normal serum calcium level is also consistent with HPT. Secondary HPT, in contrast, is diagnosed when elevated PTH levels are present in the setting of hypocalcemia caused by a known separate etiology such as chronic renal failure.
Imaging
Preoperative localization of the affected gland is important as it determines the operative approach for parathyroidectomy. If a single enlarged or hyperfunctioning gland is detected, the operation can be performed as a minimally invasive, or unilateral, parathyroidectomy (MIP). Cervical ultrasound (US) can aid both in the diagnosis of parathyroid adenomas and preoperative localization of the diseased gland. Parathyroid adenomas are recognized on ultrasound as a small, round, generally symmetrical, hypoechoic structure (Fig. 6.2). Neck ultrasound is noninvasive, relatively inexpensive, and radiation and sedation can be avoided, but the effectiveness of ultrasound is operator-dependent. Operator dependency probably explains the widely variable published accuracy rates of 48–74% [3, 14]. Ultrasound is also unable to sufficiently evaluate mediastinal glands.
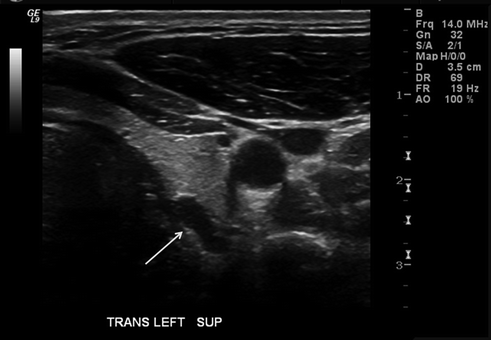
Fig. 6.2
Cervical ultrasound demonstrating a hypoechoic mass (arrow) consistent with a parathyroid adenoma posterolateral to the left thyroid lobe
One of the most common and most accurate preoperative localization studies is a dual-phase technitium-99 m sestamibi scan with single-photon emission computed tomography/computed tomography (SPECT/CT) (Fig. 6.3), with accuracy rates over 90% [3, 14, 15]. While the costs of sestamibi imaging with SPECT/CT are higher than ultrasound, this modality is less operator-dependent and is much better at detecting ectopic adenomas. However, when detection rates are based on persistent radionuclide uptake in both phase scans, the false negative rate can be as high as 40%. Recent studies including both adult and pediatric patients show that review of the early phase sestamibi scan by an experienced endocrine surgeon can result in increased preoperative localization of parathyroid adenomas, thus increasing the possibility of performing a MIP [16, 17].
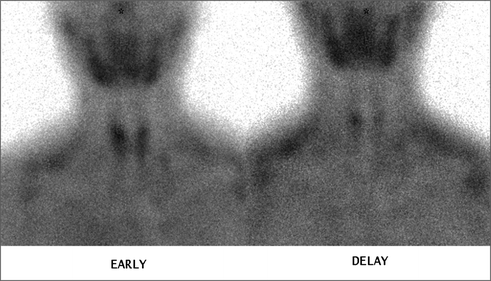
Fig. 6.3
Dual-phase technetium-99 m sestamibi scan showing increased focal uptake of the radionuclide that persists in the late phase scan, indicating a positive localization of a right side parathyroid adenoma
Additional imaging options include CT or magnetic resonance imaging (MRI). CT images rely on the vascularity of parathyroid glands and their relative enhancement with contrast compared to the surrounding structures. CT has a sensitivity of 40–86% depending on the technique and experience of the radiologist [18]. Recently the sensitivity of CT has improved to 88% with the introduction of four-dimensional CT (4D-CT) that utilizes changes in the perfusion of contrast over time in addition to the three-dimensional images [18]. In MRI imaging, hyperfunctioning parathyroid glands show contrast enhancement on T1-weighted images. Sensitivity for adenoma detection with this modality is 69–88% [18] and it may be preferred in pediatric patients, as there is no associated ionizing radiation and the costs are comparable with sestamibi SPECT/CT [3, 18].
Indications for Operation
The indications for operation vary based on the cause of hyperparathyroidism. In general, published indications derive from primary HPT in adult patients, as this is the most common type of HPT and the disease is uncommon in children. Any patient with elevated or inappropriately normal PTH and symptoms of hypercalcemia, including nephrolithiasis, nephrocalcinosis, renal dysfunction, osteopenia, pathologic fractures, osteitis fibrosa cystica, and altered neurologic function with obtundation, delirium, or coma, should undergo a parathyroidectomy [3, 18].
Regarding patients with asymptomatic PHPT, a National Institutes of Health (NIH) consensus conference published guidelines in 1990 that were amended in 2002 and 2008 [19] that propose surgery should be performed in the following circumstances:
Serum calcium is more than 1 mg/dL (>0.25 mM/L) above the upper limits of normal.
Glomerular filtration rate of <60 ml/min (per 1.73 m [2]). Below this level, elevations in serum PTH occur, and pathophysiological abnormalities associated with declining renal function may negatively influence the hyperparathyroid state.
Reduced bone density, with a Z-score of −2.5 or less in patients younger than 50 years old, or any previous fracture or fragility.
Age less than 50, as evidence supports a greater risk of complications from PHPT in these individuals over time [19].
Based on these recommendations, all children, adolescents, and young adults diagnosed with PHPT should undergo the appropriate operation to remove the affected gland(s).
If the patient’s workup is suspicious for parathyroid carcinoma and the disease appears to be resectable, any biopsy including fine-needle aspiration should be avoided, as this can violate the capsule and increase the risk for tumor implants. The patient should proceed to operative resection, as described below [4].
The indications for operative intervention in secondary hyperparathyroidism are less clear. Medical therapy is the first-line treatment for SHPT, with the goal to increase available serum calcium and vitamin D, decrease hyperphosphatemia, and increase the sensitivity of CaSRs to serum calcium. Medical therapy with calcitriol supplementation and phosphate binders is often sufficient to maintain normal PTH and phosphorous levels. Pilot studies of cinacalcet, a calcimimetic that allosterically activates CaSRs, showed that a single dose predictably lowered serum PTH, calcium, and phosphorous in pediatric renal dialysis patients, which suggests this compound might have further use as medical treatment for pediatric SHPT [20]. Although in most cases calcium and PTH levels return to normal after return of normal renal function surgical treatment of SHPT should be considered in the following circumstances:
A calcium ∙ phosphate product >70
Severe bone disease and pain
Pruritus
Extensive soft tissue calcification with tumoral calcinosis
Calciphylaxis [21].
While the vast majority of hyperparathyroidism in the setting of CKD resolves within 6 months of renal transplantation, tertiary hyperparathyroidism (TPHT) develops in 2–3% of patients who receive a renal transplant [22]. Contrary to SHPT, the primary treatment of THPT is surgical. After receiving a kidney transplant patients with SHPT are routinely monitored for resolution of their HPT. Surgical treatment should be considered if any of the following occur after renal transplantation:
Severe hypercalcemia (>11.5 mg/dL)
Persistent hypercalcemia (>10.2 mg/dL more than three months after transplant)
Severe osteopenia
Symptomatic HPT (fatigue, pruritus, bone pain, pathologic bone fracture, peptic ulcer disease, mental status changes)
Hypercalcemia with a history of renal calculi [23]
Management
Preoperative Management
The only curative management for PHPT is surgical removal of the diseased parathyroid gland(s). In general, no additional treatment is needed prior to parathyroidectomy. However, as described above, patients with SHPT and THPT are managed medically to control the levels of serum PTH, calcium, and phosphorous, and surgery is only performed if patients meet the indications presented above.
Hypercalcemic Crisis
Occasionally, patients present in hypercalcemic crisis with extremely high levels of serum calcium (generally above 14 mg/dL), oliguria or anuria, and changes in mental status [24]. This is more common in patients with NSHPT, in part because the syndrome is rare and the initial symptoms go unrecognized. Hypercalcemic crisis is a medical emergency, and patients need to be stabilized before they can undergo a parathyroidectomy. If this is the initial presentation of the patient for hypercalcemia (i.e., they have not previously been diagnosed with HPT), they should undergo a shortened diagnostic workup while they are being medically stabilized. This workup should include:
History and physical examination
X-rays of the head, thorax, vertebral column, pelvis, and long bones to look for osteolytic lesions associated with PHPT, metastases, other neoplasms
Ultrasound examination of the abdominal organs to exclude hepatic, pancreatic, renal, or gynecologic tumors
Laboratory studies including phosphate, potassium, creatinine, urea, alkaline phosphatase, complete blood count, and PTH [24].
While this workup is taking place, treatment for the severe hypercalcemia should begin. These patients are usually severely dehydrated, and the first step of therapy is intravenous hydration with normal saline. In addition, any medications that are associated with or adversely affected by hypercalcemia should be discontinued. Once intravascular volume is normalized then urinary excretion of calcium is encouraged by additional intravenous fluid. Loop diuretics are added to inhibit calcium reabsorption in the kidney and prevent fluid overload. Patients with renal failure may need urgent hemodialysis. Once the etiology of hypercalcemia is determined to be PHPT, the best definitive treatment is prompt surgical intervention. If surgery cannot be performed urgently, calcium can also be lowered with administration of gallium nitrate, bisphosphonates, or calcitonin, all of which act to inhibit osteoclasts and/or slow bone resorption [3]. However, due to the slow onset of therapeutic effect and long half-life of bisphosphonates, their use can lead to postoperative hypocalcemia, and in general they are not recommended when urgent surgery can be performed. If they are used, it is suggested that, due to children’s highly sensitive response to bisphosphonates, only half of the normal dose should be given in the acute setting [25]. Case studies and small pilot studies indicate that cinacalcet, a calcimimetic, may be used in the pediatric population in the short term to lower calcium [20, 26]. Once patients are stabilized, they can proceed to surgery.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


