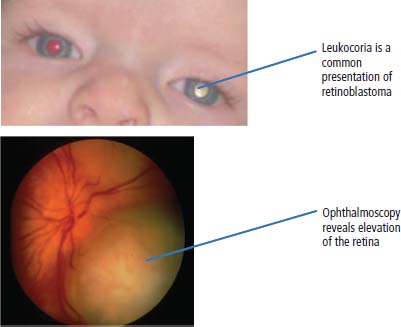
34 Cancer is a leading cause of death in children in England and Wales, second only to accidental injury. It is responsible for around 10% of deaths in childhood. In 2010, 1603 children were diagnosed with cancer in the United Kingdom and 252 died from cancer. Cancer in children is nonetheless relatively rare, affecting 1 in 600 children and includes a different spectrum of cancers than adults (Tables 1.1 and 34.1). Whilst leukaemias account for 30% of cancers in childhood, the solid tumours encountered in childhood are often embryonal in origin, and many are associated with an inherited predisposition. There are few areas of medicine that can rival the advances made in paediatric oncology in the second half of the 20th century. Eight in ten children with cancer are now cured, compared with fewer than three in ten in the 1960s. It was estimated that in 2012, 33,000 young adults in Britain aged 16–40 years are survivors of childhood cancer. Many paediatric tumours are associated with recognized familial predispositions that are due to inherited mutations of tumour suppressor genes and therefore are inherited as autosomal dominant traits. Examples are hereditary retinoblastoma (mutations of the RB gene on chromosome 13q14) and familial Wilms’ tumours (mutations of the WT1 gene on chromosome 11p13). In contrast, environmental oncogenic factors have been less readily identified for paediatric solid tumours; one example, however, is the excess of papillary thyroid cancers in children following the nuclear explosion at Chernobyl (see Chapter 2). There are a few examples of viral infections contributing to the pathogenesis of childhood solid cancers including the association between Epstein–Barr virus and endemic Burkitt’s lymphoma and Hodgkin’s lymphoma. Many solid cancers of childhood differ notably from tumours of adulthood even those affecting the same organs, whilst lymphomas and germ cell tumours in children share many features with the same illnesses occurring in adults. Embryonal tumours arise in tissues that are normally only found in the developing embryo. There are six main types of embryonal tumours: Table 34.1 Relative frequency and 5-year survival for paediatric cancers Tumours in the brain and CNS occur throughout childhood and are the second most common group of cancers in children. The age at diagnosis of these tumours is: In contrast to adult brain tumours, most (60%) are infratentorial and 75% are midline, involving the cerebellum, midbrain, pons and medulla. The most common tumours, accounting for 45%, are astrocytomas of varying grades. They include optic nerve gliomas, which are usually well differentiated tumours. A further 20% are medulloblastomas, a small round cell tumour of childhood of primitive neuroectodermal origin. Medulloblastomas usually arise in the posterior fossa and may seed metastases in the neuraxis by dropping them down the subarachnoid space into the spinal canal. Craniopharyngiomas make up 5–10% of CNS tumours of childhood and cause raised intracranial pressure, visual defects and pituitary dysfunction: usually reduced growth hormone, thyroid-stimulating hormone (TSH), antidiuretic hormone (ADH: diabetes insipidus) or luteinizing hormone/follicle-stimulating hormone (LH/FSH) abnormalities (precocious puberty or delayed secondary sexual characteristics). Suprasellar calcification is a characteristic X-ray finding. A further 1–2% are pineal region tumours that present with Perinaud’s syndrome (failure of conjugate upward gaze). Histologically, most pineal tumours are extragonadal germ cell tumours (teratomas and germinomas). Naturally, the management of these children will be determined both by the histological diagnosis and the anatomical location of the tumour and frequently involves surgery, radiotherapy and (occasionally) chemotherapy. The overall 5-year survival rates according to the histology are shown in Table 34.2. Table 34.2 The 5-year survival rates for paediatric CNS tumours Lymphomas account for 11% childhood cancers and are twice as common in boys. Hodgkin’s lymphoma accounts for 45% childhood lymphomas and the remainder are mostly high-grade non-Hodgkin’s lymphomas including Burkitt’s lymphomas. Endemic Burkitt’s lymphoma in Africa is always associated with Epstein–Barr virus present within the lymphoma cells and cases are confined to geographic areas where malaria is endemic suggesting a role for chronic immune stimulation by Plasmodium falciparum in the pathogenesis. Figure 34.1 Small blue round cell tumours. The paediatric soft tissue sarcomas account for 6% childhood cancers and include in order of frequency: rhabdomyosarcoma, Ewing sarcoma, Askin tumours and peripheral neuroectodermal tumours (PNET) (Figure 34.1). Bone tumours account for a further 4% with some overlap with the soft tissue sarcomas. Osteosarcoma is the most common bone tumour in children. Rhabdomyosarcoma is the most common paediatric soft tissue sarcoma, although only 60 children are diagnosed with this tumour in the United Kingdom each year and most are under 10 years old. Rhabdomyosarcoma may be divided into alveolar (25–30%), embryonal (50–60%) and pleomorphic (5%) variants. The embryonal type occurs in the first decade, most often in the head and neck and genitourinary tract (Figure 34.2). The alveolar type occurs in adolescents, in the forearms and trunk. The pleomorphic type occurs in adults. Consistent chromosomal translocations have been found in a number of soft tissue sarcomas, both benign and malignant. These chromosomal rearrangements may be of help diagnostically; for example, 75% of alveolar rhabdomyosarcomas harbour the t(2:13)(q35:q14) chromosomal translocation that fuses the PAX3 (Paired box 3) gene and the FKHR (forkhead) gene. The consequence of many of these translocations is the transcription of chimeric mRNA, containing 5′ sequences of one gene and 3′ sequences from another gene, and translation to hybrid proteins. Many of the genes involved with these translocations are themselves transcription factors and it is postulated that the consequence of these translocations is the aberrant expression of a number of downstream genes. Rhabdomyosarcomas present as masses that grow and may become hard and painful. Approximately 15% have metastases at presentation; most frequently in the lungs, bones and lymph nodes. Treatment involves both surgery and chemotherapy but is risk stratified, with radiotherapy reserved for those at higher risk of relapse in order to save those at low risk of recurrence from the late effects of radiotherapy. The 5-year overall survival is 75%. Figure 34.2 Embryonal rhabdomyosarcoma. Ewing’s sarcoma is named after Dr James Ewing, who described the tumour in the 1920s. Ewing’s sarcoma is a childhood bone malignancy of uncertain cellular origin that is associated with the t(11;22) chromosomal translocation that juxtaposes the EWS and Fli-1 genes, resulting in a hybrid transcript from these two transcription factor genes. This same chromosomal translocation occurs in PNETs and Askin lung tumours, suggesting a possible common origin. PNETs are thought to arise from peripheral autonomic nervous system tissue and immunostain for neuron specific enolase (NSE) as well as S-100. Morphologically, all three tumours are small round blue cell tumours – a group that also includes embryonal rhabdomyosarcoma, non-Hodgkin’s lymphoma, neuroblastoma and small cell lung cancer. About 30 children each year in the United Kingdom develop Ewing’s sarcoma. It most frequently occurs in the teenage years. Ewing’s sarcoma is very rare in African and Asian children and is not associated with familial syndromes or prior radiotherapy. It usually starts in a bone at the diaphysis or, less frequently, the metaphysic – most commonly in one of the bones of the hips, upper arm or thigh (see Figures 1.13 and 36.5), although it can also develop in soft tissue. The most common symptom is pain and swelling, but systemic symptoms such as pyrexia, weight loss and night sweats may also occur. X-rays usually demonstrate ill-defined medullary destruction, small areas of new bone formation, periosteal reaction and soft tissue expansion. Approximately a fifth of patients have metastases in their lungs or bones at presentation. Multimodality treatment including surgery, radiotherapy and chemotherapy is a standard practice for Ewing’s sarcoma, and the 5-year survival rate is 64%. The incidence of bone tumours is the highest during adolescence, although they only represent 3% of all childhood cancers. Only about 30 children develop these tumours each year in the United Kingdom. Most tumours occur in areas of rapid growth in the metaphysis near the growth plate, where cellular proliferation and remodelling are the greatest during long bone growth. The most active growth plates are in the distal femur and proximal tibia (see Figure 36.4). These are also the most common sites for primary bone cancers. Known risk factors include hereditary retinoblastoma, Li–Fraumeni syndrome and prior radiotherapy. Most primary bone tumours present as painful swellings that may cause stiffness and effusions in nearby joints. Occasionally, tumours present as pathological fractures. The radiological appearances are a lytic or sclerotic expansile lesion associated with a wide transition zone, cortical destruction, a soft tissue mass, periosteal reaction and calcification. The clinical management of bone tumours requires a specialist multidisciplinary unit including orthopaedic surgeons, plastic surgeons and oncologists. Clinical management should happen in the context of an adolescent oncology unit, since the majority of patients fit into this age group, with all its special needs. Neoadjuvant chemotherapy plays an important role in localized osteosarcoma and Ewing’s sarcoma to shrink the tumour and hopefully allow limb sparing surgery without increasing relapse rates. Postoperative adjuvant chemotherapy and radiotherapy are useful in some tumours. The 5-year survival has steadily risen from under 20% in the late 1960s to over 60%. Neuroblastoma is the most common malignancy in infants under a year old and often is clinically apparent at birth. About 100 new cases of neuroblastoma are diagnosed each year in the United Kingdom. Tumours often have amplification of the n-Myc oncogene on chromosome 1, either as small “double minute” (DM) chromosomes or as “homogenously staining regions” (HSR). They may arise from any site along the craniospinal axis derived from neural crest. The sites include abdominal sites (55%) such as the adrenal medulla (33%), pelvis (25%), thorax (13%) and head and neck (7%). In the case of head and neck neuroblastoma, these occur most commonly in the sympathetic ganglion or olfactory bulb (the latter are more common in adults). The most common finding is a large, firm and irregular abdominal mass that characteristically crosses the midline. Neuroblastomas may present with non-specific symptoms such as weight loss, failure to thrive, fever and pallor, especially if widespread metastases are present. Seventy per cent are disseminated at diagnosis via lymphatic and haematogenous spread. Metastases to bones of the skull are common, and orbital swelling is a frequent presentation. Paraneoplastic opsoclonus (rapid involuntary eye movements) or myoclonus (brief involuntary muscle twitches) is a rare feature. These tumours have the highest spontaneous regression rate of any tumour, usually by maturation to ganglioneuroma. Plain abdominal X-ray may show calcification: this occurs in 70% of neuroblastoma and 15% of Wilms’ tumours (Figure 34.3). Other diagnostic investigations include 131-I-labelled meta-iodobenzyl guanidine (MIBG) scan and blood or urinary catecholamines including vanillylmandelic acid (VMA), serum neuron-specific enolase (NSE) and ferritin. Localized disease has a high cure rate with surgery and radiotherapy. On account of the high rate of disseminated disease at presentation, which is 70%, however, the overall 5-year survival rate for neuroblastoma is 64%. Figure 34.3 Neuroblastoma. This highly malignant embryonal tumour of the kidney is the most common malignant lesion of the genitourinary tract in children. It was named after Dr Max Wilms, who first described it, and is also known as nephroblastoma. Most occur in children under 5 years old and some are hereditary. Only about 75 children develop a Wilms’ tumour each year in Britain. Wilms’ tumours are associated with congenital abnormalities including aniridia (absence of the iris usually affecting both eyes) and the WAGR syndrome (Wilms’ tumour, aniridia, gonadoblastoma and mental retardation), Denys–Drash syndrome (Wilms’ tumour, male pseudohermaphroditism and diffuse glomerular disease) and Beckwith–Wiedemann syndrome (organomegaly, hemihypertrophy, increased incidence of Wilms’ tumour, hepatoblastoma and adrenocortical tumours). Wilms’ tumours present in usually healthy children as abdominal swellings with a smooth, firm, non-tender mass. A quarter has gross haematuria and occasionally children present with hypertension, malaise or fever. Up to 20% have metastases at diagnosis; lungs are the most common sites of metastases. The mainstay of treatment is surgical resection with adjuvant radiotherapy to the tumour bed, reserved for children with a high risk of relapse. The 5-year survival for Wilms’ tumours now exceeds 80%, and one of the goals of more recent trials is to reduce the long-term morbidity of treatment. Retinoblastoma most often occurs in children under 5 years old and in a third of cases is bilateral. There are about 40 new cases of retinoblastoma diagnosed each year in the United Kingdom. Up to 40% are hereditary due to germ line mutations of the retinoblastoma (RB) gene and these children frequently have bilateral retinoblastoma and present at a younger age. Hereditary retinoblastoma was the basis of Knudsen’s two-hit model of tumour suppressor genes (Figures 1.3 and 2.14). These tumours present with whitening of the pupil, squint or secondary glaucoma (Figure 34.4). Retinoblastoma is usually confined to the orbit, and hence the cure rate with enucleation is high. Smaller tumours may be treated with localized cryotherapy, laser treatment or a radioactive iodine plaque stitched to the outer surface of the eye. Overall, 99% of children with retinoblastoma are cured. Hereditary retinoblastoma, however, is also associated with other malignancies, especially osteosarcoma, soft tissue sarcoma and melanoma. Genetic counselling is an integral part of therapy for retinoblastoma. All siblings should be examined periodically: DNA polymorphism analysis may identify relatives at high risk. Figure 34.4 Leukocoria (white eye). Fewer than 10 children in the United Kingdom develop liver tumours each year. Liver cancers are divided into hepatoblastoma (80%), which usually occurs before the age of 3 years, and hepatocellular cancers (20%), which occur at any age. Hepatoblastoma occurs as part of the Beckwith–Wiedemann syndrome and is also associated with familial adenomatous polyposis. Hepatoblastoma is the third most common intra-abdominal malignancy in young children – after neuroblastoma and Wilms’ tumours. It most frequently affects the right lobe of the liver, and 10% have disseminated disease at presentation with regional lymph node involvement or lung metastases. Hepatocellular carcinoma is associated with hepatitis B and C infection, tyrosinaemia, biliary cirrhosis and α1-antitrypsin deficiency. Surgical resection with or without neoadjuvant chemotherapy has dramatically improved the prognosis in hepatoblastoma, where the 5-year overall survival is now 70%. In contrast, the prognosis for hepatocellular carcinoma in children is not greatly different from that for adults with 5-year overall survival rates of around 25%. Langerhans’ cell histiocytosis (LCH), previously known as histiocytosis X, may not strictly be a cancer but may behave in an aggressive fashion and is often treated by oncologists. About 30 children develop LCH in the United Kingdom each year; most of them are under 2 years old. LCH is a proliferation of epidermal histiocytes or Langerhans’ cells, which are antigen-presenting dendritic cells named after Paul Langerhans, who first described them in 1868 when he was a 21-year-old medical student in Berlin. LCH comprises three overlapping syndromes: Letterer–Siwe disease occurs mainly in boys under 2 years old; Hand–Schüller–Christian syndrome has a peak of onset in children aged 2–10 years; whilst solitary eosinophilic granuloma occurs in those aged 5–15 years. Solitary eosinophilic granuloma occurs at any site in bones and is usually asymptomatic and frequently an incidental finding. Patients with Hand–Schüller–Christian syndrome often present with recurrent episodes of otitis media and mastoiditis or with polyuria and polydipsia due to diabetes insipidus. Letterer–Siwe disease presents with symptoms suggestive of a systemic infection or malignancy with a generalized skin eruption, anaemia and hepatosplenomegaly and other protean manifestations (Table 34.4). This eponymous classification of LCH has in part been abandoned, and a simpler classification into either restricted LCH (skin or bone lesions) or extensive LCH (visceral organ involvement) has been introduced. The diagnosis is confirmed histologically; the characteristic cytological features are CD1a surface antigen expression and Birbeck granules that are tennis racket shaped cytoplasmic organelles seen under the electron microscope. Localized bone disease is treated surgically or, less frequently, with radiotherapy, whilst systemic disease requires chemotherapy with cladribine often with desmopressin for the management of the diabetes insipidus. Survival exceeds 95% in unifocal disease and 80% in multifocal bone disease, but is only 50% in patients with systemic multiorgan disease. Bearing in mind the multiple manifestations and rarity of LCH, it comes as no surprise that it is the final diagnosis in an episode of House. Table 34.3 Small blue round cell tumours. Table 34.4 Clinical manifestations of extensive Langerhans’ cell histiocytosis Although many of the delayed effects of chemotherapy and radiotherapy in children are similar to those in adults, the effects on developing organs also produce unique late side effects, particularly on the skeleton, brain and endocrine systems. These delayed effects of multimodality therapy on the developing child are substantial and the late sequelae cause considerable morbidity in this group of patients where the long-term survival rates are high. Radiotherapy retards bone and cartilage growth and causes intellectual impairment, gonadal toxicity, hypothalamic and thyroid dysfunction as well as pneumonitis, nephrotoxicity and hepatotoxicity. Proton beam radiotherapy delivers charged protons (p+), the subatomic particle that is composed of 2 up and 1 down quarks, that forms the nucleus of a hydrogen atom and was discovered by Ernest Rutherford in 1920. Proton therapy offers an advance in the treatment of childhood malignancies particularly in those tumours where the delivery of high-intensity focussed radiation is critical. For example, the treatment of brain tumours using protons allows radiation to be delivered without scatter to neighbouring critical structures such as the optic chiasm in the course of irradiating the pineal gland. Late consequences of chemotherapy include infertility, anthracycline-related cardiotoxicity, bleomycin-related pulmonary fibrosis and platinum-related nephrotoxicity and neurotoxicity. Up to 5% of children cured of this cancer will develop a second malignancy as a consequence of an inherited cancer predisposition or the late sequelae of cancer treatment. Second malignancies occur most frequently following combined chemotherapy and radiotherapy. Case Study: The boy who fell off his bike.
Paediatric solid tumours
Epidemiology
Pathogenesis
Presentation and management of paediatric solid tumours
Cancer
Proportion of childhood cancers
5-year overall survival
Leukaemias
30%
83%
Brain and central nervous system tumours
27%
71%
Lymphomas
11%
88%
Soft tissue sarcomas
6%
67%
Sympathetic nervous system tumours (neuroblastomas)
5%
64%
Renal tumours
5%
84%
Carcinomas and melanomas
4%
Bone tumours
4%
61%
Gonadal and germ cell tumours
3%
92%
Retinoblastomas
3%
100%
Hepatic tumours
1%
66%
Presentation and management of CNS tumours
Tumour
5-year survival
Any paediatric CNS tumours
56%
Low-grade glioma
80%
High-grade glioma
25%
Optic glioma
80%
Brainstem glioma
5–50%
Medulloblastoma
60%
Ependymoma
60%
Pineal germinoma
90%
Pineal teratoma
65%
Craniopharyngioma
90%
Presentation and management of lymphomas
Presentation and management of soft tissue sarcomas and bone tumours
Rhabdomyosarcoma
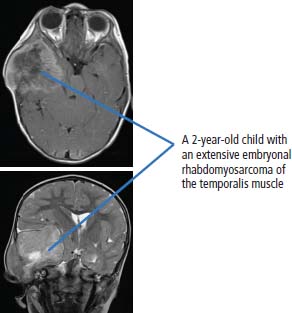
Ewing’s sarcoma
Osteosarcoma
Presentation and management of neuroblastoma
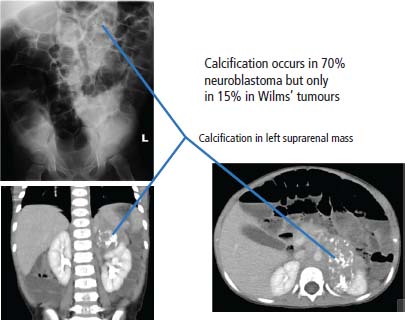
Presentation and management of Wilms’ tumours (nephroblastoma)
Presentation and management of retinoblastoma
Presentation and management of liver tumours
Langerhans’ cell histiocytosis
Paediatric embryonal tumours
Neuroblastoma
Medulloblastoma
Rhabdomyosarcoma
Wilm’s tumour (nephroblastoma)
Retinoblastoma
Hepatoblastoma (anaplastic form only)
Teenage young adult tumours
Ewing’s sarcoma/PNET
Synovial sarcoma
Adult tumours
Carcinoid tumour
Small cell lung cancer
Small cell lymphoma
System
Clinical manifestations
Systemic effects
Pyrexia, weight loss, fatigue
Bone
Painful swelling (skull (50%), femur (17%), ribs (8%), pelvis, vertebrae), associated soft tissue swelling (proptosis, mastoiditis and deafness, gums)
Skin
Scaly, erythematous, seborrhoea-like brown to red papules (behind the ears and in the axillary, inguinal and perineal areas) (50%)
Endocrine glands
Diabetes insipidus (20%) due to involvement of the hypothalamus or pituitary stalk
Bone marrow
Pancytopenia
Lymph nodes
Lymphadenopathy (30%)
Liver and spleen
Hepatosplenomegaly
Gastrointestinal tract
Failure to thrive, malabsorption, diarrhoea, vomiting (5–10%)
Lungs
Dyspnoea, honey-comb lungs, bullae, spontaneous pneumothorax, emphysema (20%)
Central nervous system
Progressive ataxia, dysarthria, intracranial hypertension, cranial nerve palsies (10%)
Complications of childhood treatment of cancer
 ONLINE RESOURCE
ONLINE RESOURCE
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


