Fig. 4.1
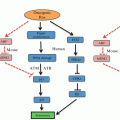
The central part of the figure represents the p53 protein indicating its main domains: the N-terminal transactivation domain, the proline-rich domain, a central DNA binding core and a C-terminal region that includes the tetramerization and the regulatory domains. Multiple residues clustered at the N- and C-termini of p53 that are post-translationally modified by phosphorylation (circles), acetylation (lozenges), ubiquitination (hexagons), methylation (squares), sumoylation (triangle), and NEDDylation (pentagons) are also shown. The histogram of p53 missense mutations (IARC TP53 Database R17, November 2013) highlights that the hotspot mutations correspond to the DNA binding core of the protein (Petitjean et al. 2007)
Although p53 acts primarily in the nucleus as a transcription factor, transcription-independent activities of p53 in the cytosol promoting apoptosis and inhibiting autophagy have been described (Green and Kroemer 2009).
4.2.2 p53 Regulation
In the absence of stress, p53 is maintained at very low levels due to continuous degradation through the ubiquitin-dependent proteasome pathway. p53 is targeted by numerous E3 ubiquitin ligases, of which Mdm2 plays the most prominent role. Mdm2 keeps p53 activity in check in different ways: Mdm2 associates to the N-terminus of p53, concealing its transactivation domain, and targets C-terminal lysines of p53 for post-translational modification such as ubiquitination and neddylation. Ubiquitination promotes proteasomal degradation and neddylation results in impaired transcriptional activity (Oliner et al. 1993; Moll and Petrenko 2003; Xirodimas et al. 2004) (Fig. 4.2). Mdm2 knockout mice are embryonic lethal due to excessive p53-induced apoptosis and this phenotype is rescued by simultaneous deletion of p53, highlighting the critical role of Mdm2 in the regulation of p53 activity (Moll and Petrenko 2003). p53 is ubiquitinated by nearly a dozen other E3 ligases, including Pirh2, COP1, ARF-BP1 (Lee et al. 2012).
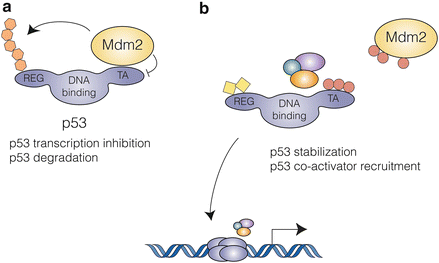
Fig. 4.2
The p53-Mdm2 interaction: (a) In unstressed cells, Mdm2 keeps p53 activity in check by two mechanisms: concealment of its N-terminal transactivation domain and targeting to ubiquitin-dependent proteasomal degradation. (b) Multiple stress activated signaling pathways result in post-translational modification of both p53 and Mdm2, disrupting their interaction and leading to p53 accumulation and activation
Mdm2 is in turn regulated by p14ARF, a tumor suppressor encoded by an alternative reading frame at the p16INK4A/ARF locus. p14ARF is induced in response to sustained oncogenic signals and protects p53 from Mdm2-dependent degradation by binding to the p53-Mdm2 complex and inhibiting Mdm2 ubiquitin ligase activity (Honda and Yasuda 1999).
MdmX (or Mdm4) is an Mdm2-structural related protein that also binds to the N-terminal domain of p53, blocking its transcriptional activity, and whose deletion produces an embryonic lethal phenotype that is rescued by loss of p53. However, unlike Mdm2, MdmX does not function as an E3 ubiquitin ligase (Marine and Jochemsen 2005).
Finally, p53 protein levels are also regulated by ubiquitin-independent proteasomal degradation (Asher et al. 2005) and through translational control (Halaby and Yang 2007).
Upon a variety of stress types, p53 is released from its negative regulators, accumulates in the nucleus and regulates the expression of numerous target genes.
4.2.3 p53 Activation
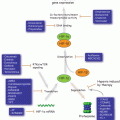
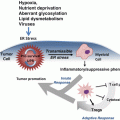
p53 integrates signals from multiple stress-activated pathways that are triggered in response to oncogene activation, DNA damage, telomere erosion, ribonucleotide depletion, altered mitochondrial and ribosomal biogenesis, nutrient deprivation, hypoxia, and loss of cell contacts (Vousden and Lane 2007) (Fig. 4.3). This integration is mediated by extensive covalent post-translational modification of both p53 and its regulators. p53 contains conserved residues that are modified by phosphorylation, acetylation, methylation, mono- and poly-ubiquitination, sumoylation, neddylation, ADP-ribosylation and glycosylation (Gu and Zhu 2012). The vast majority of these modifications are induced following stress, in a stimuli- and tissue-specific manner; only a few are present in unstressed cells and are removed upon p53 activation.
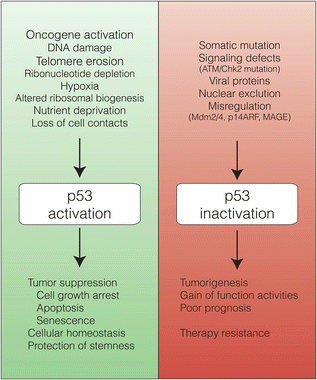
Fig. 4.3
A great variety of cellular stimuli activate p53 that, through the regulation of the expression of multiple target genes, elicits numerous cellular processes responsible of preventing tumorigenesis and ensuring cellular homeostasis. In contrast, inactivation of the p53 pathway or somatic mutation of the p53 protein leads to tumorigenesis and is associated with poor prognosis and therapy resistance. Mut-p53 often exhibits GOF activities that contribute to the tumoral phenotype
Numerous studies have reported roles for these modifications in the regulation of almost every aspect of p53 biology: stability, conformation, DNA binding, cellular localization, and protein-protein interaction. However, their absolute requirement for p53 activation has been a source of debate. Mutation studies in mice suggest that there is a significant degree of redundancy among p53 modifications, particularly in the case of phosphorylation. This is reflected in the number of phosphorylation sites, the number of residues phosphorylated by a single kinase, and the number of kinases phosphorylating a single residue. This redundancy might ensure a robust signaling to p53 by stress-activated pathways. Alternatively, it might represent a way to fine-tune the p53 response (Dai and Gu 2010).
Two of the best studied modifications of p53 are phosphorylation at serine 15 and serine 20. Ser15 and Ser20 phosphorylation by DNA damage- and stress-activated kinases (ATM/ATR, Chk1/Chk2, DNA-PK, p38, MAPK, JNK) reduces the interaction of p53 with Mdm2 and enhances the recruitment of transcriptional co-activators (Kruse and Gu 2009). Phosphorylation of Mdm2 by some of the same kinases ensures the disruption of its interaction with p53, possibly as a safety mechanism (Meek and Knippschild 2003) (Fig. 4.2).
p53 is acetylated at multiple C-terminal lysines by p300, CBP (CREB Binding Protein), and other histone acetyltransferases. This modification has been shown to (1) stabilize p53 by competing with ubiquitination of the same C-terminal lysines, (2) enhance sequence-specific DNA binding and transcriptional activity in tissue culture systems, (3) release promoter-bound p53 from a repressed state mediated by Mdm2 and Mdm4 binding and (4) contribute to selectivity in target gene transcription (Kruse and Gu 2009; Dai and Gu 2010). p53 function is therefore regulated by an equilibrium between acetylation and deacetylation, which is maintained by histone deacetylase 1 (HDAC1) and Sirtuin 1 (SIRT1). SIRT1 expression is elevated in many types of cancers and p53 deacetylated at lysine 382 by SIRT1 has a significantly reduced ability to induce apoptosis (Luo et al. 2001).
4.3 Tumor-Suppressive Activity of p53
Appropriate p53 activation is crucial for tumor suppression. This notion is highlighted by different observations, the first of which came from cell biology experiments showing that wild type p53 (wt-p53) can counteract oncogene-induced fibroblast proliferation (Hermeking and Eick 1994). Then, a number of clinical studies evidenced that p53 is mutated in more that 50 % of human tumors (http://www-p53.iarc.fr/) and that patients with Li-Fraumeni Syndrome bearing a mutated-p53 (mut-p53) allele are highly cancer-prone (Malkin 2011). Concluding evidence for a master role of p53 in tumor suppression came from animal models: all mice carrying deleted p53 alleles develop cancer (Purdie et al. 1994; Jacks et al. 1994; Donehower et al. 1992).
The following points of this section underline the molecular mechanisms involving the p53 pathway as a guardian against early events of oncogenesis. Such mechanisms include well-known activities controlling cell proliferation as well as novel mechanisms comprising metabolism regulation and control of stem cells.
4.3.1 A Barrier for Oncogenesis
Oncogene expression boosts cell cycle progression and consequently the activation of p53. This activation depends on the p14ARF/Mdm2 pathway. Oncogene-induced p14ARF expression inhibits Mdm2 E3 ubiquitin-ligase activity and therefore p53 stability (Llanos et al. 2001). In fact, a marked percentage of tumor cells show deficiencies in p14ARF expression or in p53 activity (Honda and Yasuda 1999). If the p53 pathway is intact, oncogene activation triggers the p53 response. Activated p53 will then induce the expression of a variety of target genes involved in cell cycle arrest, senescence or cell death. However, recent data emphasize the relevance of p53 as a regulator of metabolism to exert its full activity as a tumor suppressor (Gu and Zhu 2012).
4.3.1.1 Regulation of Cell Proliferation
A well described p53-dependent way to sustain cell cycle arrest is through the expression of the cdk inhibitors p21 (el-Deiry et al. 1993) and 14-3-3 sigma (Laronga et al. 2000). Oncogene activation can also induce replicative senescence through the p53/PML axis (Pearson et al. 2000). PML itself is a target of p53 (de Stanchina et al. 2004), relocalizes p53 to the PML nuclear bodies causing p53 acetylation and activation, and triggering senescence (Bischof et al. 2002; Pearson et al. 2000).
Depending on cell type and persistence of the stimuli, p53 can efficiently remove potentially dangerous cells by triggering the apoptotic cell death process; an extreme way to eliminate cells with permanent defects considered unfeasible to overcome. In fact, p53 activates a significant set of genes able to induce apoptosis, suggesting that this process is one of the key tools for p53 to eradicate cells with oncogenic potential. Pro-apoptotic genes directly induced by p53 include Bax and Bid, members of the Bcl-2 family (Miyashita and Reed 1995; Sax et al. 2002), PUMA (p53 upregulated modulator of apoptosis) (Nakano and Vousden 2001), NOXA (NADPH oxidase activator) (Oda et al. 2000), and Apaf-1 (apoptotic peptidase activation factor) (Moroni et al. 2001) that activate mitochondrial apoptosis. Also, p53 is able to promote apoptosis by inducing death receptors as Apo1/Fas (Muller et al. 1998), DR4 (Liu et al. 2004) and KILLER/DR5 (Wu et al. 1997).
Therefore, activation of selected p53 target genes involved in the control of cell cycle, senescence or apoptosis is critical to prevent cell transformation (Fig. 4.3). However, more recent findings linking p53 to glucose metabolism, redox balance and autophagy have opened a new window to understand the tumor suppressive activity of p53.
4.3.1.2 Promotion of Normal Cell Metabolism
Glucose Metabolism
Metabolism of tumor cells is different from that of normal cells mainly regarding glucose consumption. Tumor cells uptake and metabolize large amounts of glucose to generate lactate in order to obtain energy, even in normal aerobic conditions (Warburg effect). In this situation, normal cells produce ATP molecules through the more efficient tricarboxylic acid cycle coupled to oxidative phosphorylation. Active glycolysis in tumor cells could allow growth under hypoxia before new blood vessels begin to form in the tumor mass (angiogenesis). However, it seems that tumor cells adopted glycolysis also in normoxic conditions for a fast, instead of efficient, energy production. In fact, activation of oncogenic pathways has been shown to promote the tumor cell metabolic program (glycolysis). The PI3K/AKT pathway is activated by nutrients and promotes cell growth. This pathway is frequently hyperactivated in cancer cells and is also involved in cancer cell metabolism (Vousden and Ryan 2009). In addition, the oncogenic transcription factors c-MYC and HIF regulate genes that accelerate glycolysis (Wise et al. 2008; Yeung et al. 2008), suggesting that metabolic reprogramming is required for cell transformation. Interestingly, regulation of cell metabolism has been included among the p53 tumor-suppressive functions.
p53 can be activated by metabolic stress resulting from low oxygen availability and limited nutrient or energy (Vousden and Ryan 2009). Recent findings have shown that p53 plays a crucial role in controlling glucose metabolism. Activated p53 can regulate the expression of glucose transporters GLUT1, GLUT4 and also GLUT3 (through the NF-kB pathway) (Schwartzenberg-Bar-Yoseph et al. 2004; Kawauchi et al. 2008) and other components of glucose metabolism such as hexokinase II HK2 (Mathupala et al. 2006), TIGAR (TP53-induced glycolysis and apoptosis regulator) (Bensaad et al. 2006) and PGM (phosphoglycerate mutase) (Kondoh et al. 2005). Conversely, p53 induces the SCO2 (cytochrome c oxidase 2) (Matoba et al. 2006) and AIF (apoptosis-inducing factor) genes (Stambolsky et al. 2006) to contribute to oxidative phosphorylation. In this way, p53 promotes normal cell metabolism and ATP production through oxidative phosphorylation and reduces the influx of glucose.
Redox Balance
Reactive oxygen species (ROS) are normally produced during general metabolism and, when tightly controlled, they actually promote cell proliferation. However, high levels of ROS cause protein, DNA and lipid oxidation and are associated with aging, cardiopathology and cancer. In order to maintain a correct cellular environment, p53 induces a set of antioxidant proteins (Ladelfa et al. 2012) such as glutathione peroxidase GPX1 (Tan et al. 1999), superoxide dismutase (MnSOD) (Drane et al. 2001), TP53INP1 (TP53-induced protein 1) (Cano et al. 2009) and members of the sestrin family, Sesn1 and Sesn2 (Budanov et al. 2004). In contrast, induction of p53-dependent apoptosis has a strong oxidative component since most of the p53 targets that induce apoptosis through the intrinsic mitochondrial pathway produce high levels of ROS (Ladelfa et al. 2012). In summary, p53 appears to play an important role in promoting normal cell metabolism by regulating glycolysis and the oxidative balance. Importantly, a mut-p53 unable to induce apoptosis due to lack of key lysines, maintained its ability to regulate metabolism and displayed tumor suppressive function (Gu and Zhu 2012) stressing the relevant role of p53 in controlling metabolism.
Autophagy
Another metabolic process in which p53 is involved is autophagy (macro-autophagy), a way to obtain energy through controlled degradation of organelles and proteins into lysosomes when nutrients are scarce. Therefore, through the autophagic process, cells replenish energy reserves and promote cell survival. p53 plays a dual role in this process, by inducing autophagy from the nucleus and repressing it in the cytoplasm (Comel et al. 2014). The main pathway controlled by p53 is the AMPK-mTOR axis. This pathway inhibits autophagy and, in turn, p53 inhibits the pathway at multiple levels. p53 was shown to activate transcription of several genes whose protein products can directly or indirectly regulate signaling from AMPK to mTOR: TSC2, beta-1 and beta-2 subunits of AMPK and sestrins 1 and 2 (Feng et al. 2005, 2007; Budanov and Karin 2008). In addition, DRAM (damage-regulated autophagy regulator), another p53 target gene, is a lysosomal protein that activates autophagy, linking this process to apoptosis (Crighton et al. 2006). In unstressed cells, p53 has a cytoplasmic component that counteracts autophagy. Forced depletion of p53 or cell starvation, which physiologically induces p53 degradation, promotes autophagy. This mechanism was observed only in cells bearing wt-p53 and is associated to downregulation of the key autophagy protein LC3. By playing this role p53 could balance the energy requirements both when the autophagy rate is low or high (Maddocks and Vousden 2011; Scherz-Shouval et al. 2010).
All the above mentioned data point to p53 as pivotal protein able to sense a wide range of stimuli to trigger responses focused on preserving the proper cell function. Once this balance is broken, the p53 response becomes focused on the elimination of potentially dangerous cells.
4.3.2 p53 Controlling Stemness
In addition to the aforementioned role in differentiated cells, p53 is also involved in the protection of stem cells (SCs). SCs are undifferentiated cells capable of self-renewal and generating specific cell lineages through asymmetrical cell division. Embryonic stem cells (ESCs) can differentiate into ectoderm, mesoderm and endoderm lineages, while adult stem cells (ASCs), which are found in many tissues, regenerate the corresponding tissue-specific cells. This hierarchical and one-way order puts SC at the top, followed by somatic cells with diverse degree of differentiation. However, breakthroughs in the SC field showed the possibility of inducing pluripotent SC (iPSCs) from differentiated mouse embryonic and adult fibroblasts, through the ectopic expression of four proteins: Oct3/4, Sox2, KLF4 and c-Myc (Takahashi and Yamanaka 2006).
Earlier studies showed that the classical p53 response is reduced in mouse ESCs (Rogel et al. 1985; Sabapathy et al. 1997; Han et al. 2008; Aladjem et al. 1998). This behaviour presents a paradox, given the high cell division rate of SCs. Many recent studies shed light on this field indicating that p53 response in SCs is responsible for suppression of self-renewal and induction of differentiation when genomic stability is compromised after DNA damage (Aloni-Grinstein et al. 2014). Activated p53 in SCs binds to the promoters and represses the expression of Nanog and Oct4, two well-known markers of SCs and key proteins for the maintenance of the self-renewal and undifferentiated state. Thus, upon DNA damage, repression of these two genes by p53 forces SCs differentiation (Lin et al. 2005). In-deep studies of p53 in SCs identified a number of target genes involved in development and differentiation such as those belonging to the FOX, SOX, TBX, CBX and homeodomain families (Akdemir et al. 2014; Morey and Helin 2010).
Mesenchymal SCs (MSCs) are a type of ASC that generate different mesodermal cells when they differentiate. p53 point mutations were found in aged MSCs along with embryonic markers, suggesting that non functional p53 could be related to tumor development of mesenchymal origin (i.e., sarcoma) during ageing (Li et al. 2007). Transformation or dedifferentiation of ASCs can lead to the formation of cancer stem cells (CSCs). These cells are reservoirs of cancer cells with similar features (self-renewal and generation of more differentiated tumor cells) to that of normal SCs (Aloni-Grinstein et al. 2014). Using mouse models of MSCs, it was demonstrated that alterations in the p53 pathway but not the retinoblastoma (pRb) pathway is associated with sarcomatogenesis (Rodriguez et al. 2012; Rubio et al. 2010) and that fibrosarcomas derived from aged animals could be originated from a MSCs bearing mut-p53 (Li et al. 2007).
4.4 Deficient p53 Activity in Tumor Cells
It is now clear that defects in any step of the p53 pathway can facilitate tumor promotion. In addition to being a key protein in the control of carcinogenesis, proper p53 activation is also an important aspect of cancer therapy. Radiotherapy and chemotherapy rely mainly on the induction of (selected) DNA damage-induced apoptosis or at least on inhibiting cell proliferation. Therefore, induction of p53 by ionizing radiation (radiotherapy) or DNA-damaging agents (chemotherapy) mostly correlates with good therapy outcome.
In ovarian cancer patients, p53 alterations correlate with resistance to platinum-based (cisplatin or carboplatin) chemotherapy, early relapse and shortened overall survival (Reles et al. 2001; Shelling 1997). Non response to purine analogs therapy (fludarabine or pentostatin) and poor survival were reported in chronic B-cell leukemia patients with p53 gene deletion (Dohner et al. 1995). p53 mutations are also associated with poor response to chemotherapy and radiotherapy in colorectal and gastric cancer patients (Hamada et al. 1996). For other cancers, resistance to therapy due to non functional p53 activity is not well established and seems to depend on both cellular context and chemotherapeutic drugs.
Inadequate p53 function in tumor cells is mainly caused by gene mutation, but it has been also shown that oncogenic proteins can target p53 activity (Fig. 4.3). That knowledge obtained from basic cancer research was essential to develop p53-based therapies. Both scenarios exhibit a degree of complexity and are described here.
4.4.1 Mutant p53 in Tumors
A small number of tumors have no detectable p53 protein expression due to frameshift or nonsense mutations. However, approximately 75 % of tumor-associated alterations in p53 are missense mutations, with a single amino acid change in the p53 protein. These mutants are expressed at very high levels in cancer, suggesting that their expression can confer advantages to tumor cells (Petitjean et al. 2007). Indeed, 20 years ago, Dittmer and co-workers reported that the introduction of mut-p53 into p53 null cells results in a new phenotype and suggested that mut-p53 can gain a novel transforming function (Dittmer et al. 1993). Since then, several cell culture assays have demonstrated that different mut-p53 proteins acquire gain of function (GOF) activities such as promotion of cell survival, drug resistance, anchorage-independent growth, increased colony formation and genomic instability among others (Muller and Vousden 2014) (Fig. 4.3).
Consistent with p53 functioning primarily as a transcription factor, the vast majority of the missense mutations observed in p53 lie in the core DNA binding domain. Mutations frequently found in p53 (hotspot mutations) can be classified as class I/“DNA contact” mutations (R248, R273) or class II/“conformational” mutations (R175, R245, R249, R282) that disrupt or destabilize p53 structure (Brosh and Rotter 2009; Petitjean et al. 2007; Muller and Vousden 2013) (Fig. 4.1).
Mice models were developed in order to study mut-p53 GOF in a physiological context, allowing more properly assessment of cancer parameters such as metastatic potential, spectrum of tumor generation, angiogenesis and tumor latency. Knock-in mice in which the endogenous p53 mouse allele was replaced with orthologs of specific human hotspot p53 mutants, such as p53 R172H/− (corresponding to human p53R175H) and p53 R270H/− (corresponding to human p53R273H), developed a broadened spectrum of tumor and showed increased metastatic potential compared to p53 −/− mice (Lang et al. 2004), supporting the notion of a GOF mechanism for these mutants.
Similar studies were performed in a humanized p53 knock-in (HUPKI) mouse model (Luo et al. 2001). Of special interest, p53hupki R248Q/− mice showed the most potent mut-p53 GOF reported to date, displaying diminished survival and decreased tumor latency compared to p53 −/− mice (Hanel et al. 2013).
Lessons learnt from cell culture assays and knock-in mouse models support the notion that different mut-p53 promote different oncogenic responses, not only by losing wt-p53 function but also by gaining novel pro-oncogenic functions.
4.4.2 Inactivation of Wild Type p53
In addition to mutations, p53 can also be inactivated by oncogenic viral proteins encoded by small DNA viruses. In fact, p53 was discovered as a 53 kDa protein complexed to the SV40 large T viral oncoprotein (LT) (Lane and Crawford 1979). LT directly binds the p53 DNA binding domain, thus blocking p53 interaction with DNA (Ali and De Caprio 2001). High risk human Papilloma Virus E6 oncoprotein interacts with E6AP (E6 associated protein) and p53 to promote ubiquitin-mediated degradation of p53 (Huibregtse et al. 1993; Scheffner et al. 1990). In adenovirus transformed cells, p53 inhibition of cell proliferation and induction of apoptosis are blocked by the E1B-55K protein. This protein binds with high affinity to the p53 transcriptional activation domain, probably sterically blocking interactions of p53 with its coactivators (Berk 2005).
Currently, growing evidence suggest that, similar to mut-p53 proteins, viral oncoproteins-p53 complexes could contribute to tumorigenesis through GOF mechanisms. According to this, new transcriptional and biological activities, such as the activation of the IGF-I signalling pathway by the LT-p53 complex, have been reported (Bocchetta et al. 2008).
Another line of attack to wt-p53 comes from cellular proteins highly expressed in tumor cells. For example, high levels of Mdm2 are commonly observed in human cancers due to gene amplification, elevated transcription, increased mRNA stability, enhanced translation, and altered post-translational modifications (Riley and Lozano 2012). High levels of Mdm4 protein are also found in a variety of human cancers mostly due to Mdm4 gene amplification (Markey 2008). Conversely, loss of p14ARF, which antagonizes Mdm2, allows tumor development without loss of wt-p53 (Stott et al. 1998).
Analysis of tumors and tumor cell lines that retain wt-p53 pointed to nuclear exclusion as yet another mechanism of tampering with p53 function, particularly in breast carcinomas and neuroblastomas (Moll et al. 1992, 1995). Although Mdm2 amplification was not observed in those samples, subsequent studies showed that Mdm2 might play a significant role in the cytoplasmic p53 phenotype (Lu et al. 2000).
Finally, the tumor-specific MAGE-I (Melanoma antigen gene) proteins, especially Mage-A proteins, negatively regulate p53 activity possibly by both direct binding to p53 (Marcar et al. 2010) or by recruiting HDACs (Monte et al. 2006) or KAP1 (Doyle et al. 2010; Yang et al. 2007) proteins to p53-containing complexes (Ladelfa et al. 2011). As MAGE-I proteins are expressed in humans cancers of different lineages, interfering with its expression or function could have an impact on the treatment of a wide range of human cancers, especially those harbouring wt-p53.
4.5 p53, a Key Target for Cancer Therapy
Many aspects of p53 biology put it at the center of cancer therapy treatments. As mentioned earlier, loss of p53 function is the most frequent alteration in human cancer, highlighting the unquestionable role that p53 plays in tumorigenesis. At the same time, loss of p53 also contributes to therapy resistance. Drug dose escalation is one strategy to improve the therapeutic outcome in p53-negative tumors but this also enhances the toxicity in normal tissues, in particular in those that are highly sensitive to p53 induced apoptosis, generating chemotherapy side effects. In summary, p53 is both at the origin of the disease and a challenge to its treatment. In view of all this, p53 represents an extremely attractive and highly strategic target for cancer treatment.
4.5.1 Proof-of-Principle and Strategy to Restore p53 Function
A key question arises before pursuing any p53-based cancer therapy. Once mutation or inactivation of p53 has led to tumor formation, will restoring p53 function be sufficient to stop its progression or even promote tumor regression?
Elegant mouse models were engineered to answer this question that provided promising proof-of-principle results. Through different mechanisms, mainly apoptosis or senescence, re-expression of endogenous p53 was able to cause stasis or regression of the established tumors (Martins et al. 2006; Ventura et al. 2007; Xue et al. 2007). These observations bolstered research on multiple approaches to reinstate p53 function in tumor cells and hundreds of clinical trials looking into or targeting p53 are currently ongoing (http://clinicaltrials.gov) (Kenzelmann Broz and Attardi 2010; Hoe et al. 2014).
p53 deficiency in human cancer can be classified into three categories: loss of p53, mutation of p53 and inactivation of wt-p53. As previously discussed, p53-null tumors should be considered separately as missense mutations of p53 can have a substantially different effect on cellular responses compared to deletion or nonsense mutations. Anti-cancer therapeutic strategies based on targeting p53 can similarly be divided into corresponding categories: activation/re-introduction of wt-p53 and reactivation of mut-p53 (Frezza and Martins 2012).
4.5.1.1 Activating Wild Type p53
Current p53 gene therapy relies mostly on p53 delivery by replicative-deficient adenoviruses (Ad-p53), which had shown dramatic apoptotic responses and tumor regression in cell culture and rodent models. Since 2003, Ad-p53 Gendicine (Shenzhen SiBiono GeneTech, China) is being applied in China in combination with radiation therapy for the treatment of head and neck cancer and hepatocellular carcinoma. Ad-p53 Advexin (INGN-201), developed by Introgen Therapeutics, is yet to be approved by the U.S. Food and Drug Administration and several clinical trials are underway to test the efficacy of this and other Ad-p53 (SCH-58500), alone, in combination therapy, or following surgery (Kim and Dass 2011).
Attenuation of the p53 response in tumors expressing wt-p53 can result from alterations in p53 regulatory proteins. Basic research conducted over the past 20 years has singled out Mdm2, as well as Mdm4, as the major negative regulators of p53 with oncogenic potential. The efficient and natural strategy to activate p53 shared by p14ARF, some ribosomal proteins or the DNA damage-induced post-translational modifications, consists in disrupting the Mdm2-p53 interaction. Based on this, the use of small molecule compounds, antisense therapy and stapled peptides committed to interfere with Mdm2-p53 complex is a key strategy currently explored in order to activate p53 (Wade et al. 2013).
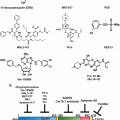
X-ray crystallography of the p53-Mdm2 interface combined with site-directed mutagenesis experiments and studies using peptidic inhibitors showed that their interaction was a suitable drug target and allowed to derive pharmacophore models for this protein-protein interaction (Chene 2004; Shangary and Wang 2009). Together with high throughput screenings, these models were used in structure-based de novo design and computational 3D screenings of the National Cancer Institute database to identify hundreds of potential Mdm2 inhibitors (Essmann and Schulze-Osthoff 2012) (Table 4.1). RG7112 (Hoffman-La Roche), a Nutlin 3a potent derivative, and the spiro-oxindole MI-219 are some of these small molecule compounds that have progressed into advanced preclinical development or early phase clinical trials.
Table 4.1
p53 targeting and p53-based therapies