and Paul Telfer2
(1)
Department of Haematology, Guy’s and St Thomas’ Hospital, London, UK
(2)
Department of Haematology, Royal London Hospital, London, UK
Introduction
In this section, we describe the structure and function of the haemoglobin molecule, red blood cells and blood vessels. This is not a comprehensive account, but will help in understanding the principles as they relate to sickle cell disease.
Basic Physiology
Hemoglobin
The hemoglobin molecule is composed of four subunits each of which contains a protein chain (globin), which encloses a flat ring-like molecular structure (the heme prosthetic group) where oxygen is bound. Hemoglobin has been refined through evolution and is the central player in a system of molecules, biochemical reactions and biological compartments whose integrity is essential for gas exchange. The process involves uptake of oxygen in the alveolar beds of the lungs, where concentration (measured as partial pressure) is high, and release in the tissues, where concentration is low. Simultaneously, carbon dioxide is taken up in the tissues and released in the lungs.
When oxygen binds to a heme subunit, there is change in the shape of the hemoglobin molecule, due to interactions with amino acids in the adjacent globin chain. This facilitates binding at the other heme subunits within the tetramer and promotes oxygen carriage. These interactions are affected by amino acid substitutions in the globin chain.
Hemoglobin is compartmentalized within the red blood cell and if released into the circulation as free hemoglobin is not able to participate in effective gas exchange. Free hemoglobin may cause circulatory and renal toxicity, through binding and inactivating nitric oxide, a small molecule which has an important role in controlling blood flow (principally through its effects on the vessel wall).
This compartmentalization of hemoglobin within the red cell also enables it to play a major role in carbon dioxide transport, through buffering hydrogen ions produced from the conversion of carbon dioxide and water to bicarbonate and hydrogen ions.
2,3-biphosphoglycerate (2,3-BPG) is an important by-product of the breakdown of glucose (glycolysis) within the red blood cell and interacts with hemoglobin to facilitate oxygen delivery by shifting the oxygen dissociation curve to the right. When oxygen concentration in the tissues is low, and metabolism of glucose are predominantly anaerobic rather than aerobic, excess lactic acid is produced, and this results in increased intra-erythrocytic 2,3 DPG.
Hemoglobin is synthesized in red cell precursors (erythroblasts) during their differentiation within the bone marrow, and continues to be synthesized in the cytoplasm of the youngest population of circulating red cells, the reticulocytes. Mature red cells lack the enzymes and organelles required for its continued synthesis.
Iron destined for heme synthesis is transported to the erythron in its oxidized (ferric, Fe 3+) state bound to plasma transferrin (Tf). Erythroblasts express plentiful transferrin receptors on their surface, and these can bind the iron-Tf complex, leading to internalization by endocytosis. Intracellular mobilization of iron occurs through a pathway consisting of acidification of the interior of the endosome, release and reduction of iron to its ferrous (Fe2+) state, and transportation of Fe2+ across the endosomal membrane into the cytoplasm and subsequently into the mitochondrion via the transport protein mitoferrin. Within the mitochondrion, Fe2+ iron is finally incorporated into heme as the last step of heme biosynthesis, in a reaction catalysed by the enzyme ferrochelatase. Heme is then transported back into the cytoplasm for incorporation into hemoglobin.
Molecular Genetics
The beta globin cluster on Chromosome 11 consists of an array of genes encoding the beta–like globins. The alpha globin gene cluster is physically separated on chromosome 16. It differs in its genetic organization, but is regulated in concert with the beta globin cluster to ensure co-ordinated expression of the correct genes. Sequential expression of alpha-like and beta-like genes at different stages of development ensures that specific hemoglobin is produced according to the needs of the developing embryo, fetus or infant. Fetal hemoglobin is the predominant hemoglobin from about 12 weeks of gestation up until birth. It consists of two alpha and two gamma globin chains (HbF, α2γ2) and has enhanced oxygen affinity compared to adult hemoglobin (HbA, α2β2). This ensures that oxygen is transported efficiently across the placental membrane from HbA containing maternal red cells into HbF containing fetal red cells.
The control of expression of the globin gene clusters during development has been the subject of sustained genetic research over the past four decades. The incentive has been partly because globin gene expression is seen as a paradigm for the understanding of gene expression. Also, research has informed therapeutic approaches to managing hemoglobin disorders through gene therapy or reactivation of developmental globin gene expression in order to circumvent the genetic changes affecting adult hemoglobin (HbA).
The Sickle Mutation
The mutation is a C to A substitution at codon 6 of the beta globin gene, which results in a substitution of the neutral amino acid valine for glutamic acid. The sickle hemoglobin molecule is designated HbS (α2βs 2).
The Sickling Process
In the deoxygenated state, HbS molecules form a polymeric structure where individual molecules stack up into fibres, and then aggregate into a rope-like structure of inter-twined fibre-pairs. When hemoglobin becomes oxygenated, the array denatures. The structure is formed though non-covalent interactions between adjacent hemoglobin molecules. This is made possible through exposure of binding sites during the process of oxygenation and deoxygenation of HbS. Molecules are stabilized in this polymeric structure because of a new interaction between the substituted β6 valine and a neutral pocket on adjacent hemoglobin molecules. The normal glutamic acid residue at position 6 of beta globin cannot participate in this interaction because of its shape and charge (Fig. 1.1).
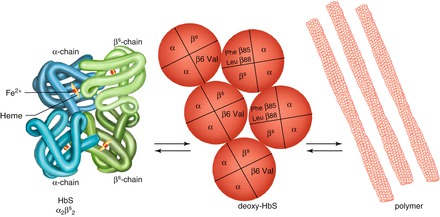
Figure 1.1
The sickling process. The hemoglobin molecule, comprising four globin chains each with a heme group, is shown on the left. The sickle mutation results in a valine substitution at position 6 of the beta globin chain. This is able to form a new interaction with amino acids at positions 85 and 88 of adjacent beta globin chains when hemoglobin is in the deoxygenated state. Multiplication of these interactions leads to propagation of sickle hemoglobin polymers
The time lag from deoxygenation of hemoglobin to polymer formation enables the red cell to traverse the capillary bed and return to the lungs for uptake of oxygen, whereupon the polymer can deaggregate. This sickling/desickling cycle is repeated with each circulation of the red cell. If retarded, extensive polymerization of hemoglobin can occur before returning to the alveolar beds, resulting in red cell damage. The potential for a damaging positive feedback of more obstruction leading to more sickling can easily be appreciated.
Several factors influence the speed of polymer formation:
1.
Intracellular hemoglobin concentration. Higher mean corpuscular hemoglobin concentration (MCHC) facilitates sickling. Lower MCHC (e.g. with co-inheritance of alpha thalassemia, or concomitant iron deficiency) retards sickling.
2.
Presence of other intracellular hemoglobins. Some hemoglobins (e.g. HbA, HbF) inhibit sickling, and some hemoglobin variants (HbC, HbDPunjab, HbOArab) are permissive. These interactions explain why carriers of sickle cell (HbAS) and double heterozygotes with hereditary persistence of fetal hemoglobin (HbS/HPFH) are protected against sickling, while some double heterozygotes (e.g. HbSC, HbSDPunjab, HbSOArab) have a clinically significant sickle condition.
3.
Intracellular pH and temperature. Low pH and a raised temperature increase sickling in vitro, and may also play a role in clinical situations. These may be part of the explanation for onset of acute crisis associated with fever and conditions of acidosis.
Consequences of Sickling
Erythrocytic Damage
Intracellular sickling of hemoglobin can damage the red cell through several inter-related mechanisms, all leading to reduced red cell survival (hemolysis)
1.
Damage to the membrane and cytoskeleton. Red cells are adapted to squeeze through narrow diameter blood vessels (the narrowest capillary vessels are about one third of the red cell diameter). This is made possible by the elasticity of the protein matrix arranged underneath the cell membrane (the cytoskeleton). Extensive polymer formation and repetitive cycles of sickling and de-sickling eventually result in irreversible damage to the membrane and cytoskeleton, giving rise to irreversibly sickled cells (ISC’s). An increase in circulating ISC count is associated with enhanced hemolysis but not with frequency of vaso-occlusive crises.
2.
Changes in the red cell membrane. Damage to the cell membrane and cytoskeleton also results in the expression of adhesive cell surface molecules, and increased exposure of negatively charged phospholipid on the red cell surface, leading to enhanced adherence to the vascular endothelium.
3.
Red cell dehydration. Erythrocytes containing sickle hemoglobin, particularly reticulocytes and ISC’s, can become dehydrated, with an increase in mean intra-corpuscular hemoglobin concentration (MCHC). This is independent of the state of hydration of the body as a whole. Sickling of hemoglobin enhances loss of water and electrolytes and potentiates influx of calcium across the cell membrane. The Gardos channel, an important governor of ion and water exchange, is activated by increased intracellular calcium and mediates loss of potassium ions and water. It is known to be activated in SCD erythrocytes, and has been seen as a possible therapeutic target.
4.
Impaired anti-oxidant mechanisms. Maintenance of hemoglobin in its reduced state, and protection of the erythrocyte against the toxic effects of denatured hemoglobin and unbound heme requires an intact system of intracellular anti-oxidant pathways, and these are also impaired in SCD erythrocytes.
Vaso-Occlusion (Fig. 1.2)
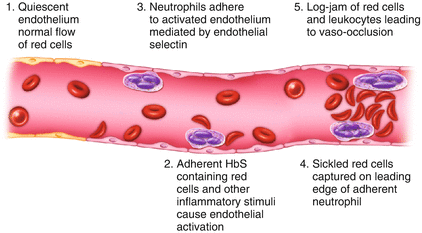
Figure 1.2
Blood cell and endothelial interactions leading to vaso-occlusion
Experimental systems have been developed to visualize the process of vaso-occlusion, making use of transgenic mice expressing exclusively human sickle hemoglobin. Vaso-occlusion can be induced by the inflammatory process which occurs when a viewing chamber is inserted surgically to observe the microvascular flow in these animals. This is greatly accentuated by injecting inflammatory cytokines such as tumour necrosis factor (TNF). These activate a whole cascade of effectors (leucocytes, endothelium, platelets, coagulation factors) implicated in the process of vaso-occlusion. An important early event is adhesion of HbS-containing erythrocytes to the vessel wall, either directly to the endothelium, or indirectly, through binding to adherent, activated neutrophils. This results in a log-jam of erythrocytes which occlude flow, leading to ischemia, infarction, and subsequent reperfusion injury (Figs. 1.2 and 1.3). Characterization of ligands and receptors involved in these adhesive interactions and the events involved in cellular activation are leading to development of anti-sickling drugs which attenuate the clinical effects of vaso-occlusion.
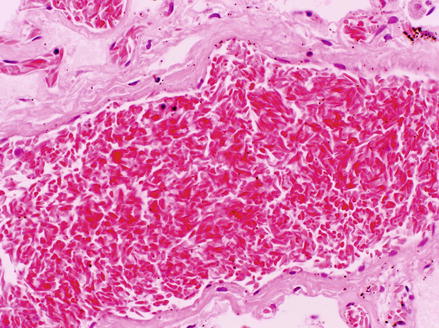
Figure 1.3
Histological specimen of the lung from a fatal case of acute chest syndrome showing occlusion of a medium sized pulmonary vessel with sickled erythrocytes
Nitric Oxide Depletion
Nitric oxide (NO) is a small molecule which has an important function in controlling local blood flow. It is produced in the endothelium from the amino acid L-arginine in a reaction catalysed by nitric oxide synthase (NOS). One form of this enzyme catalyses a constant background production, whilst a further inducible form catalyses a higher rate of production in response to endothelial activation. NO diffuses from the endothelium into underlying smooth muscle cells and causes relaxation of smooth muscle, vasodilatation and increased blood flow.
Depletion of NO is an important cause of circulatory disturbance and vascular damage in SCD. Proposed mechanisms for this include breakdown of sickled erythrocytes within the vessel lumen (intravascular hemolysis). This releases free hemoglobin to scavenge NO and also causes diversion of NO to detoxify antioxidants produced from degraded hemoglobin.
Inflammation and Tissue Damage
The pathway to tissue damage in SCD is likely to involve endothelial activation, adhesion and activation of leukocytes, together with free-radical generation from interaction of NO with free plasma hemoglobin, release of heme from degraded intravascular hemoglobin, and hypoxia-reperfusion injury. Figure 1.4 illustrates how these mechanisms can interact and form a positive feedback loop, leading to more vaso-occlusion and more tissue damage.
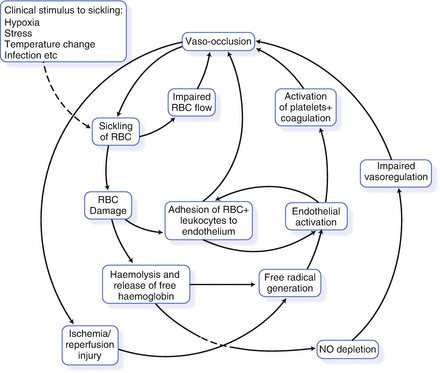
Figure 1.4




Pathophysiological processes involved in vaso-occlusion
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
