Fig. 11.1
Schematic illustrating the cellular organization in the human adult lung. (a) The architecture of the proximal airway epithelium in the trachea, bronchi, and bronchioles. (b) The airway epithelium in the distal airways, including the terminal bronchioles (left, straight) and alveoli (right, curved). The key indicates the diverse cell types of the lung epithelium
Adult stem cells contribute to the maintenance and repair of mature tissues through (1) self-renewal and (2) the generation of more differentiated progenitor or effector cells. By examining cellular recovery following targeted lung injury, a large number of studies have uncovered a group of progenitor cells with stem cell properties that are important for regenerating the lung epithelium (Rock and Hogan 2011). The prevailing view is that distinct pools of stem or progenitor cells exist along the proximal-distal axis, including separate stem/progenitor compartments in the proximal airways, bronchioles, and alveoli. In a contrasting model, a single stem cell has been proposed to have the capability of self-renewal and differentiation into all epithelial cell types in the lung (Kajstura et al. 2011). The same cells have also been proposed to give rise to endothelium within the pulmonary vasculature, although this finding remains controversial.
Genetic Regulation of Self-Renewal and Implications for Cancer
Stem cell numbers in normal tissues are tightly regulated, whereas in many tumors there is an expansion of cells with stem-like properties. Activation of known oncogenes can disrupt the normal homeostatic regulation of (1) cell death and/or (2) cell differentiation. For example, Bcl-2 overexpression within HSCs results in reduced cell death through inhibition of apoptosis (Domen et al. 2000), and c-myc overexpression prevents the differentiation of HSCs (Prochownik and Kukowska 1986). These studies illustrate how stem cell numbers during tumorigenesis are affected by an imbalance in the regulatory networks governing self-renewal.
Many of the signaling pathways implicated in the regulation of self-renewal in normal tissues are emerging as key players in tumorigenesis. Classic examples are the Wnt, Notch and Sonic hedgehog (Shh) pathways. Wnt encompasses a large family of secreted proteins that bind to receptors to activate β-catenin. Wnt/β-catenin signaling plays a pivotal role in the self-renewal of normal stem cells and in the oncogenic progression of many human cancers (Holland et al. 2013). The Notch family of receptors controls development and differentiation processes in many tissues. Aberrant Notch signaling is a common occurrence in cancer, and Notch inhibition is now being explored as a promising therapeutic strategy for solid cancers as well as leukemias (Purow 2012). Shh is a secreted molecule that binds to the Patched family of receptors. Shh signaling regulates self-renewal in a number of normal tissues. Mutations in Patched-1 are observed in basal-cell carcinoma and medulloblastoma, and pharmacological inhibition of the Shh pathway has recently shown great promise for treatment of these diseases (Rudin et al. 2009; Sekulic et al. 2012). Taken together, these observations indicate that stem cells and cancer cells often rely on similar self-renewal pathways. Further inquiry will be necessary to exploit these pathways specifically in cancer cells without adversely affecting normal developmental programs.
Stem Cells and Tumorigenesis
Studies on the cellular hierarchy in the normal tissues have direct implications for tumorigenesis and the cellular origin of cancer. Because stem cells possess properties that allow for perpetual self-renewal and survival, these cells may be most capable of giving rise to malignant clones (Reya et al. 2001). Indeed, this phenomenon has been demonstrated in mouse models of multiple tumor types, including in adenocarcinomas of the colon, breast, and prostate (Visvader 2011).
Distinct progenitor cell populations are thought to be capable of giving rise to subtypes of lung cancer. Lung adenocarcinoma may originate from type-II alveolar cells (Xu et al. 2012) or from bronchioalveolar stem cells situated at the bronchiolar-alveolar junction (Kim et al. 2005). Squamous cell carcinoma has been proposed to arise from tracheal basal cell progenitors (Sutherland and Berns 2010). Small cell lung cancer can arise from pulmonary neuroendocrine cells in experimental systems (Sutherland et al. 2011). Future investigations harnessing lineage tracing techniques in mouse models of lung cancer (Rawlins et al. 2009), will be needed to fully characterize the oncogenic potential of individual populations of normal stem and progenitor cells in the adult lung.
Cancer Stem Cell Theory and Lung Cancer
CSCs are malignant cells capable of self-renewal and differentiation into progeny within tumors. Owing to these properties, surviving CSCs within primary tumors or metastases can reconstitute an entire tumor following treatment or upon transplantation into immunodeficient mice (Reya et al. 2001). The first unequivocal demonstration of CSCs was in acute myeloid leukemia (Lapidot et al. 1994), and the presence of CSCs in solid cancer was first shown for breast adenocarcinoma (Al-Hajj et al. 2003). CSCs have since been identified in many different tumor types (Nguyen et al. 2012).
A number of methods have been proposed to enrich for cellular fractions containing cells with stem-like properties in lung cancer. Most of these methods are designed to prospectively identify cells that are more likely to undergo clonogenic growth under defined laboratory conditions. They discriminate between cells based on physical or biochemical properties, such as Hoechst dye efflux (Ho et al. 2007) and Aldefluor assays (Jiang et al. 2009), or based on cell surface expression of specific proteins, such as CD133 (Eramo et al. 2008) or CD166 (Zhang et al. 2012).
There are practical limitations of these assays that have important ramifications for the CSC theory. First, the degree of overlap between cellular populations identified by different assays is not well characterized. It has been demonstrated that the proper CSC enrichment strategy likely depends on the histologic or genetic subtype of NSCLC (Curtis et al. 2010). This presents obvious challenges to the application of the CSC theory to the study of inherently heterogeneous diseases such as lung cancer. Additionally, the defining property of a CSC is the ability to give rise to a new tumor that contains more CSCs as well as the more differentiated non-tumorigenic cancer cells that make up the bulk of most tumors. In light of this, rigorous studies of CSCs in NSCLC must continue to functionally evaluate CSC populations and must include in vivo assays rather than relying on surrogate markers or tissue culture assays.
The Role of Radiotherapy in the Treatment of Lung Cancer
The role of radiotherapy in the treatment of NSCLC depends primarily on stage at diagnosis.
For early-stage NSCLC, the standard-of-care treatment involves resection of the primary tumor by lobectomy with simultaneous dissection of hilar and mediastinal lymph nodes. In cases where no lymph node spread is detected at diagnosis, >60% of patients may be cured by surgery alone. However, up to one third of early stage NSCLC patients are not surgical candidates. For these patients, high-dose-per-fraction stereotactic ablative radiotherapy (SABR), also called stereotactic body radiotherapy, can achieve similar rates of cure (Senan et al. 2011). For unresectable disease that has spread to the lymph nodes in the chest, external beam radiotherapy is delivered with conventional fractionation (e.g., 2 Gy per fraction for ~30 fractions) either alone or with concurrent platinum-based chemotherapy.
Radioresistance is commonly seen with conventionally fractionated radiotherapy. Local recurrence is detected clinically in up to half of cases. Thus, the identification of novel strategies for sensitizing NSCLC cells to the effects of ionizing radiation—without increasing treatment-related morbidity—will be important for improving the outcomes for these patients. For patients treated with SABR, however, local control is approximately 90% (Senan et al. 2011). The predominant mode of failure for these patients is within lymph nodes and/or distant sites, suggesting that improved outcomes in this group of patients will require identification of individuals at high risk of disease spread who would likely benefit from treatment intensification, for example by addition of adjuvant chemotherapy or immunotherapy.
Radioresistance in Cancer
Resistance to radiotherapy is an important clinical problem because of its impact on patient survival and subsequent treatment options. The primary cytotoxic effect of therapeutic ionizing radiation (IR) is DNA damage. Double strand breaks (DSBs) are the most lethal form of DNA damage caused by IR because these are the most difficult for the cell to accurately repair. The most common type of radiotherapy in use today for lung cancer involves treatment using photons (X-rays). Photons are primarily indirectly ionizing, meaning that the damage they inflict is a result of the production of free radical reactive oxygen species (ROS) within cells.
The degree of radioresistance varies across tissue types. Contributing to this heterogeneity are the diversity of cellular architecture within an organ and the availability of molecular oxygen supplied by the vasculature. In addition, many cell intrinsic factors influence resistance to IR. Cells produce a group of antioxidant enzymes that neutralize ROS (Kobayashi and Suda 2012). The expression of these ROS scavengers has been shown to affect resistance to IR and cytotoxic chemotherapy in human cancers, presumably as a result of reduced levels of DNA damage. Once DNA damage has occurred, the ability of a cell to sense and repair it depends on the type of DNA lesion, the phase of the cell cycle, and the expression of repair machinery. In the case of catastrophic DNA damage that cannot be accurately repaired, the p53 pathway normally triggers apoptosis; however, in cancer deranged p53 signaling and/or other pro-survival genetic lesions can lead to an inability for cells to die appropriately (Helton and Chen 2007; Smith et al. 2010). Genomic instability resulting from such checkpoint failure can lead to higher mutation rates and rapid selection of resistant cells. Thus, there is a multistep progression toward cell death as a result of IR, and each step presents an opportunity for a cancerous cell to intervene and to survive treatment.
Mechanisms of Radioresistance in CSCs
Tumor recurrence following radiotherapy can have dire consequences. Retreatment presents significant challenges and the potential for increased toxicity. Moreover, the risk of metastasis generally rises once a tumor recurs. Importantly, radioresistant cells that lack the capacity for self-renewal would not be as problematic as resistant clones that have this capacity and are thereby able to initiating regrowth of a tumor (Reya et al. 2001).
Recent findings have confirmed that CSCs from multiple tumor types including lung cancer are programmed to preferentially survive the effects of chemotherapy and radiotherapy (Diehn et al. 2009a; Jiang et al. 2009). Mechanisms of CSC resistance to IR have begun to be uncovered in human cancers and mouse models (Morrison et al. 2011) (Fig. 11.2). Both cell-intrinsic and microenvironment-dependent mechanisms have been proposed. Cell-intrinsic mechanisms include slower progression through the cell cycle, increased expression of ROS scavenging enzymes, enhanced sensing and repair of DNA damage, and pro-survival signaling.
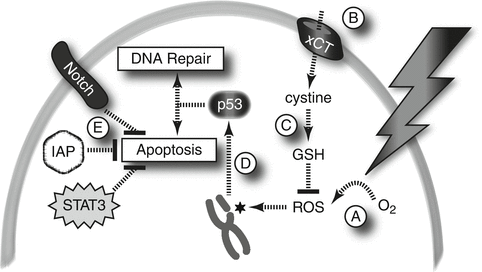
Fig. 11.2
Mechanisms of radioresistance in CSCs. (A) Hypoxia: CSCs in hypoxic microenvironments may be resistant to IR by virtue of the relative paucity of molecular oxygen from which to derive ROS. (B, C) ROS scavenging: CSCs import cystine and produce high levels of enzymes necessary to convert it into GSH, a potent ROS scavenger. (D) Checkpoint signaling: DNA DSBs that result from IR/ROS activate p53 through ATM, CHK1, and CHK2 signaling; these pathways may be more active in CSCs. (E) Survival signaling: Pro-survival signals, including XIAP, STAT3, and the Notch pathway, are activated in CSCs and lead to inhibition of apoptosis
The quiescent state of some stem cells and CSCs may render them relatively resistant to IR since they are less likely to traverse mitosis, the most radiosensitive phase of the cell cycle. However, CSCs in some tumor types have been shown to cycle at similar rates to other tumor cells (Al-Hajj et al. 2003). Radioresistance of CSCs could also be explained by lower levels of ROS as compared to non-CSC tumor cells as a result of higher levels of glutathione (GSH) and other ROS scavengers in those cells (Diehn et al. 2009b; Ishimoto et al. 2011). DNA repair in response to IR may be more robust in CSCs as a result of robust checkpoint signaling. CSCs in glioma preferentially repair DNA DSBs following IR in a manner dependent upon the Chk1/Chk2 checkpoint kinases (Bao et al. 2006). Similarly, in a mouse model of medullosblastoma, nestin-positive CSC-like cells preferentially undergo IR-induced p53-dependent cell cycle arrest and recovery, whereas nestin-negative tumor cells are more likely to undergo p53-dependent apoptotis (Hambardzumyan et al. 2008). Thus, CSCs in multiple tumor types appear to be protected against catastrophic DNA damage through a combination of ROS scavenging and checkpoint activation leading to enhanced repair of DSBs.
Recent advances have also highlighted the contributions of the tumor microenvironment to CSC radioresistance. Dysfunctional tumor vasculature and unrestrained cancer cell proliferation contribute to pockets of hypoxia where radioresistant CSCs may arise. Hypoxia is detrimental to effective cell killing by IR due to reduced ROS and correlates with radioresistance and treatment failure in human cancer (Brizel et al. 1999). In response to hypoxia, hypoxia-inducible factors (HIFs) drive gene expression programs that promote self-renewal and survival of glioma CSCs (Li et al. 2009). With these data in mind, there has been interest in sensitizing tumors to radiation through increased tissue oxygenation (Overgaard 2011), but the effect of such maneuvers on the CSC populations in clinical samples is not known.
Radioresistance in Lung CSCs
Several of the mechanisms discussed above have been shown to contribute to radioresistance of CSCs or CSC-like cells in lung cancer. For example, ROS scavengers are expressed at higher levels within CD133-positive NSCLC cells (Salnikov et al. 2010), and augmented checkpoint signaling may contribute to reduced apoptosis in response to IR (Lundholm et al. 2013). With regards to the microenvironment, direct measurements with oxygen electrodes have revealed that even small lung tumors are hypoxic, and the degree of hypoxia correlates with prognosis (Le et al. 2006).
Taken together, CSCs from various tumor types utilize multiple and perhaps distinct strategies for avoiding cell death in response to IR. Even within a single tumor, genetic heterogeneity and varied microenvironments provide opportunities for CSCs with different properties to arise (Odoux et al. 2008). Thus, for reproducible clinical benefits to be observed with novel radiosensitizers directed at CSCs, multiple radioresistance mechanisms may need to be targeted simultaneously.
Targeting CSCs in Lung Cancer
Efforts to translate the CSC hypothesis into clinical practice will drive future advances in the management of lung cancer. Strategies for interfering with resistance mechanisms of CSCs, or for otherwise leading to their selective inhibition, are discussed below.
Promoting Apoptosis in CSCs
Recent evidence supports the view that CSCs in some tumor types depend on a blockade of apoptosis pathways in order to survive the effects of therapeutic radiation (Hambardzumyan et al. 2008). The Inhibitor of Apoptosis Protein (IAP) family, key regulators of apoptosis in cancer cells, has been implicated in radioresistance of multiple tumor types including lung cancer (LaCasse et al. 2008). XIAP inhibitors block clonigenic survival of glioma CSC-like cells in vitro (Vellanki et al. 2009). Importantly, XIAP inhibitors have also been shown to increase radiosensitivity in a NSCLC cell line (Cao et al. 2004). STAT3, like XIAP, promotes cell survival in cancer. Recent reports have shown promise for a strategy of radiosensitizing CSCs with STAT3 inhibitors (Hsu et al. 2011). Further studies are needed to evaluate the effects of XIAP and STAT3 inhibition on the radiosensitivity of normal tissues.
Promoting Differentiation in CSCs
CSCs are capable of self-renewal and differentiation. Driving cellular differentiation may be an effective strategy for limiting the degree of self-renewal and potential for tumor expansion and spread. All-trans retinoid acid (ATRA) has potent effects on cellular differentiation. Preclinical investigations have demonstrated an inhibitory effect on CSCs from glioma and head and neck squamous cell carcinoma (Ning et al. 2013).
Inhibiting Self Renewal in CSCs
In addition to strategies for promoting cellular death or differentiation, signaling pathways involved in self-renewal of normal and cancer stem cells are being targeted for therapeutic gain. Clinical investigations are ongoing for Wnt, Notch, and Hedgehog inhibitors in solid tumors (Morrison et al. 2011). Inhibition of telomere reverse transcriptase (TERT) is another promising therapeutic strategy for preventing unrestrained self-renewal of CSCs (Ning et al. 2013). For all of these inhibitors, their ability to sensitize stem cells and CSCs to IR will need to be investigated.
Targeting CSC Cell Surface Proteins
Selective inhibition of CSCs through cell surface markers is an area of active investigation. Targeting of c-kit and CD133 has been investigated in lung cancer and glioma, respectively. In some NSCLC cell lines, a sub-population of CSC-like c-kit-positive cells are resistant to cisplatin but are effectively inhibited by the c-kit inhibitor, imatinib (Levina et al. 2010). Importantly, the combination of cisplatin with either imatinib or anti-SCF antibodies (SCF: stem cell factor, the c-kit ligand) had additive effects on growth inhibition. Whether additive effects are also seen with radiotherapy is not known. In glioblastoma multiforme, in which CD133 is thought to label CSCs (Singh et al. 2004), Wang et al. designed an antibody-conjugated cytotoxin that is activated by near-infrared laser light (Wang et al. 2011). The result was targeted killing of CD133-positive cells in vitro. While not immediately translatable to clinical practice, these proof-of-concept experiments suggest that the combination of antibody-based therapies with radiotherapy could result in eradication of CSCs.
Related posts:
 Decellularized Stem Cell Matrix: A Novel Approach for Autologous Chondrocyte Implantation-Based Cartilage Repair
Hypertensive Emergencies in Children After Stem Cell Transplantation: Care in Selecting Hypotensive Drugs
Emerging Concepts of Stem Cell Organization in the Normal Lung and in Lung Cancer
Decellularized Stem Cell Matrix: A Novel Approach for Autologous Chondrocyte Implantation-Based Cartilage Repair
Hypertensive Emergencies in Children After Stem Cell Transplantation: Care in Selecting Hypotensive Drugs
Emerging Concepts of Stem Cell Organization in the Normal Lung and in Lung Cancer
 Induced Pluripotent Stem Cells Differentiate into Functional Cardiomyocytes
Induced Pluripotent Stem Cells Differentiate into Functional Cardiomyocytes
 Preoptic Regulatory Factor-2, a Rhogap Domain Protein that Modifies Cell Cycle Progression and Apoptosis in the CNS
Preoptic Regulatory Factor-2, a Rhogap Domain Protein that Modifies Cell Cycle Progression and Apoptosis in the CNS
 Improved Renal Revascularization Outcomes in Pigs Using Stem Cells
Improved Renal Revascularization Outcomes in Pigs Using Stem Cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


