Fig. 6.1
Multiple immunomodulatory coinhibitory and costimulatory receptor-ligand pairs have been identified (although not all are depicted here). These pathways set the immunologic context when an antigen is presented on a T-cell receptor (TCR) to a major histocompatibility (MHC) complex
6.2 Cytotoxic T-Lymphocyte-Associated Antigen-4 (CTLA-4): A Paradigm for Immune Checkpoint Blockade
Cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4, CD152) was the first recognized and is the best characterized inhibitory immune checkpoint molecule [12, 13]. CTLA-4 is the only checkpoint currently targeted with an FDA-approved therapeutic drug, ipilimumab. The success of this drug has dramatically increased interest in cancer immunotherapy generally and immune checkpoint blockade more specifically. Many lessons have been learned in the journey from CTLA-4 discovery until efficacy was proven for ipilimumab, not the least of which is that immune checkpoint modulation can treat cancer in a clinically meaningful way. During the development of CTLA-4 blocking monoclonal antibodies (mAb), much has been learned about dosing, toxicity, combination therapy, and tumor response that are now and will continue to be useful as other immune checkpoint therapies are developed.
6.2.1 CTLA-4 Function
When CTLA-4 (CD152) was first reported in 1987, it was presumed to play a role in controlling T-cell activation given its close sequence homology with CD28, its proximity to CD28 on chromosome 1, and its expression on cytotoxic T-lymphocytes (CTLs) coinciding with T-cell activation [12]. The first CTLA-4−/− knockout mice, created in the mid-1990s, confirmed that CTLA-4 played a key role in T-cell homeostasis as the mice quickly succumbed to polyclonal lymphoproliferative disease characterized by massive expansion of activated T-cells [14]. Since then, it has become clear that CTLA-4 functions as a negative counterpart to CD28, the required costimulatory signal for the activation and expansion of T-cells.
For T-lymphocytes to be activated, an antigen-specific T-cell receptor (TCR) must bind to a MHC complex containing the appropriate peptide in its binding grove. While this is necessary, it is not sufficient to complete activation. A number of additional regulatory pathways have since been elucidated that closely control T-cell activation to ensure appropriate, directed immune responses under normal circumstances. Among these pathways, costimulation with CD28 (on the T-cell) binding to B7-1 (CD80) or B7-2 (CD86) on the antigen presenting cell (APC) is perhaps the most important and best known; B7-1 and B7-2 are expressed on APCs and are typically upregulated after activation [15, 16].
As the negative counterpart to CD28, CTLA-4 is an inhibitory checkpoint molecule expressed on activated T-cells and constitutively expressed on regulatory T-cells (Treg) [13]. After TCR-antigen mediated activation of T-lymphocytes, expression of CTLA-4 on the cell membrane increases dramatically. CTLA-4 appears to suppress the immune activation through multiple pathways and the relative importance of each in overall immune homeostasis and in disease-related autoimmunity and immune suppression is not clear [17]. Additionally, the importance of CTLA-4 in long-term immune memory has yet to be fully elucidated [18]. These remain areas of active research.
The CTLA-4 receptor controls effector T-lymphocyte activation by competitive binding with CD28 as well as through internal and external signaling. CTLA-4 binds the same ligands as CD28 (B7-1 and B7-2) but with 20–100 times greater avidity and can accommodate two ligands, whereas CD28 can only bind one [19–21]. CTLA-4 appears to blunt T-cell responses by not only competitively binding the CD28 ligands, B7-1 and B7-2, but also by receptor-mediated induction of cell cycle arrest, decreasing production of IL-2, limiting T-cell dwell time, and enhancing Treg function, among other mechanisms [18]. There is evidence that competitive binding of B7-1 and B7-2 by CTLA-4 remains the most important function in counteracting CD28-mediated T-cell stimulation as treatment of CTLA-4-deficient mouse models with CTLA-4-immunoglobulin fusion protein (CTLA-4Ig) can abrogate the lymphoproliferative autoimmunity which would otherwise be fatal [22]. Additionally, the singular importance of B7-1 and B7-2 in these pathways is demonstrated by the fact that mice deficient in CTLA-4 as well as B7-1 and B7-2 do not demonstrate lymphoproliferative autoimmunity [23]. Unlike CD28 which has some level of constitutive expression on most T-cells, CTLA-4 is only expressed in significant quantity on effector T-cells after activation. CTLA-4 reaches a maximal expression level as long as 48 h after the T-cell is activated serving as a negative feedback loop to turn off or prevent an overly robust immune response as well as to prevent autoimmunity [20, 24] (Fig. 6.2).
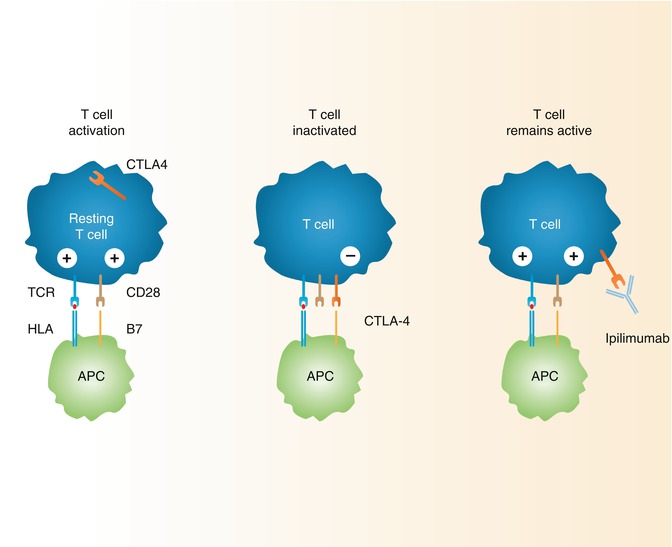
Fig. 6.2
Mechanism of action of CTLA-4 in suppressing activated T-cells and proposed mechanism of action for ipilimumab
In addition to directly and indirectly inhibiting effector T-lymphocyte activation and proliferation, CTLA-4 interacts with Tregs in a manner important to its overall function. As previously stated, CTLA-4 is expressed at some constitutive level on Treg cells, and higher levels of expression may be rapidly mobilized from an intracellular source [17]. The exact role that Treg-mediated immune suppression plays in the overall context of CTLA-mediated immune control is not entirely clear although it is an area of active research [16]. There is evidence from lymphocytes treated with anti-CTLA-4 mAbs in vitro which suggests that CTLA-4 blockade mediates the immune system by both direct activation of effector T-lymphocytes and Treg depletion, dependent on the mAb subtype and its ability to stimulate antibody-dependent cytotoxicity (ADCC) [25, 26].
The important role of CTLA-4 in Treg homeostasis and immune control has become clear in multiple experiments. Treg-mediated CTLA-4 inhibits B7-1 and B7-2 expression on dendritic cells [27]. Murine models with CTLA-4-deficient CD4+ FOXP3+ (Treg) lymphocytes developed lymphoproliferative disease [27]. Additionally, CTLA-4 plays an active role in Treg homeostasis as blocking the receptor with anti-CTLA-4 mAbs leads to a rapid proliferation in peripheral Treg cells [28–30]. This action is thought to be due to CTLA-4 counteraction against CD28-stimulated proliferation of Tregs as blocking both CTLA-4 and CD28 leads to a contraction in the peripheral Treg population [16, 28]. However, expansion of Tregs with CTLA-4 blockade does not appear to lead to increased Treg function [31]. Similarly, in murine organ transplant models, deficiency of CD28 or both B7-1 and B7-2 leads to a significant decrease in the Treg population; however, the mice get paradoxical acceleration of graft rejection inversely proportional to the Treg level [31].
As work progresses in deciphering the mechanisms of the CTLA-4 receptor’s complex interplay within broader immune homeostasis, the CTLA-4 receptor is an attractive therapeutic target with potential for revolutionizing immunotherapy for cancer as well as other disease conditions. The identified roles that CTLA-4 plays in human disease are substantial and ever-growing. There is evidence that CTLA-4 polymorphisms plays a role in autoimmune conditions such as type 1 diabetes, thyroiditis autoimmune hypothyroidism, and Graves’ disease [32–35].
6.2.2 Preclinical Development of CTLA-4 Blocking Therapy
Initial work to determine the expression patterns and function of CTLA-4 used fusion proteins of immunoglobulin with the extracellular domain of CTLA-4 (CTLA-4Ig) [36–40]. The CTLA-4 Ig protein was noted to bind to B7 with high avidity and, by competitively binding with B7 molecules, prevent CD28 mediated T-cell activation. Treatment with CTLA-4 Ig led to increased survival of transplanted xenografts, inhibition of experimental autoimmune encephalitis in murine models, and blocked antitumor immunity in murine models [36, 37, 41, 42]. Taken together, these studies supported the idea that CTLA-4 played a role in suppressing the body’s T-cell response by competitive binding. Additionally, the CTLA4 Ig fusion protein was developed into FDA-approved therapeutic drugs by Bristol-Myers Squibb (New York, NY) for treatment of rheumatoid arthritis (abatacept) and immunosuppression after renal transplant (betalcept) [43–45]. Anti-CTLA-4 mAbs were first created by Walunas et al. Their uses further supported the notion of an inhibitory role for CTLA-4 [46, 47]. The functional role of CTLA-4 was later confirmed with the aforementioned CTLA-4 knockout mice [14].
Soon thereafter, work with anti-CTLA-4 blocking mAbs for therapeutic purposes proceeded. Initial animal studies confirmed that the mAbs could indeed augment immune response to peptide antigens [47]. Not long after this, CTLA-4 mAbs were used to experimentally overcome antitumor immune tolerance. In 1996, Leach et al. reported results of experiments in mice injected with 51BLim10 colon cancer cell lines. Treatment with CTLA-4 mAbs resulted in regression of the tumors, whereas the control mice succumbed to the cancer by 35 days. Additionally, prior treatment with CTLA-4 mAbs provided protective immunity to a secondary challenge of tumor cells [48]. Additional work by Yang et al. confirmed the antitumor effect of CTLA-4 mAb in CSA1M fibrosarcoma and OV-HM ovarian carcinoma mice models and also demonstrated that the effectiveness was dependent on the stage of tumor growth, with later stages being less susceptible to mAb-enhanced eradication [49]. Since then, multiple murine models of various cancer types, specifically glioblastoma, sarcoma, breast, prostate, and colon cancer models, have proven the efficacy of anti-CTLA-4 mAb [18, 50–54]. However, in multiple other murine tumor models, including different cell lines from the same types of cancer that responded in previous experiments, anti-CTLA-4 mAb monotherapy was shown to have low or no antitumor effect [55–64]. Some have suggested, and it stands to reason, that less immunogenic tumors are less likely to respond to CTLA-4 mAb blockade alone [18]. Less immunogenic cancers may require additional antigenic stimulation, through active immunotherapy or through radiation or chemotherapy-induced cell death to realize its full potential as will be discussed further.
6.2.3 CTLA-4 Blockade Monotherapy in Melanoma
Two mAbs, ipilimumab and tremelimumab, were developed in parallel. The therapies underwent phase III trials that ultimately led to approval for ipilimumab for treating metastatic melanoma and showed disappointing results for tremelimumab.
6.2.3.1 Ipilimumab
Based on the work in murine models, fully humanized IgG1 CTLA-4 mAbs were created by Medarex, Inc (Princeton, NJ; purchased by Bristol-Myers Squibb, New York, NY, in 2009) using a transgenic hybridoma HuMAb mouse model. The proprietary mouse model has multiple genetic modifications designed to facilitate production of high-avidity human IgG mAbs [65]. The mAb used for initial in vivo testing was selected based on affinity and specificity for CTLA-4 as well as ability to block the binding site [66]. The antibody, called 10D1 (later designated MDX-010 and ipilimumab), also had cross-reactivity with macaques monkey CTLA-4. It was initially tested in this setting where it was shown to increase antibody response to hepatitis surface antigen as well as a human melanoma cell vaccine. Additionally, the macaques did not demonstrate polyclonal T-cell activation or autoimmunity [66]. Based on this work, ipilimumab proceeded with human trials.
Phase I Trials
Two initial phase I pilot studies of ipilimumab monotherapy were conducted in 2002 in castrate-resistant prostate cancer (CRPC) and unresectable malignant melanoma. In both studies, a single dose of ipilimumab was given at 3 mg/kg to each patient. This dose was selected based on pharmacokinetics from prior animal studies [43]. In the first study, 2 out of 14 patients with CPRC had a transient prostate-specific antigen (PSA) response of >50 % lasting up to 5 months [67]. In the second trial, 2 of the 17 patients with metastatic melanoma had a partial response to the treatment without any serious adverse events [67]. In 2003, a phase I trial of ipilimumab monotherapy was conducted in previously immunized metastatic melanoma and ovarian cancer patients also at a dose of 3 mg/kg. Five of the nine patients (three melanoma patients and two ovarian cancer patients) were previously immunized with autologous, irradiated, GM-CSF secreting tumor cells (GVAX), and all demonstrated some objective response to ipilimumab with some developing “extensive tumor necrosis.” Four melanoma patients previously immunized with a multiple peptide vaccine did not appear to respond. There were no serious toxicities reported [68]. From there, a series of phase I trials were conducted both in metastatic melanoma as well as prostate cancer [43]. An additional phase I trial, reported in 2003, tested ipilimumab in 11 patients with colon cancer, prostate cancer, or lymphoma at 3 mg/kg initial dose followed by three monthly doses at 1.5 mg/kg. A partial response was noted in lymphoma patients and appeared to coincide with observed autoimmune toxicities [69].
Phase I/II Trials of Ipilimumab Monotherapy in Melanoma
Based on these experiences, the drug proceeded with phase I/II trials in melanoma. Melanoma was selected as a cancer of interest for CTLA-4 blockade therapy based on observed responses in phase I trials and also its historical consideration as an immunogenic tumor based on cases of spontaneous regression and its response to IL-2 and other immunotherapies. Notable trials in this category helped determine the biologic dose used later in phase III trials. One phase I/II trial enrolled 88 patients with unresectable stage III or IV melanoma and gave them a single dose at 20 mg/kg, multiple doses at 5 mg/kg, or multiple doses of 10 mg/kg. All doses were tolerated, although more immune-related adverse events (irAE, discussed further below) were noted in the group receiving 10 mg/kg serially. A disease control rate (DCR), defined as patients with an objective response or stable disease, was 39 % (9 of 23 patients) in the 10 mg/kg serial dosing group and less than 15 % in other groups [70]. Another NIH-sponsored study enrolled 139 patients with metastatic melanoma and treated them with ipilimumab and a peptide vaccine (n = 54) or ipilimumab monotherapy with intra-patient dose escalation (n = 84). The doses were escalated from 3 mg/kg every 3 weeks to up to 9 mg/kg or until disease progression or unacceptable toxicity. An objective response rate of 17 % was observed for all patients [71]. Both trials noted significant rates of irAEs with 86 and 62 % of patients, respectively, experiencing some toxicity. Rates of ≥ grade 3 toxicity were 36 and 19 %, respectively. Of note, both trials found a positive correlation between the presence of irAEs and clinical response.
Phase II Trials of Ipilimumab Monotherapy in Melanoma
Multiple phase II trials have been conducted on ipilimumab monotherapy and ipilimumab with other agents in melanoma. The first series of phase II trials were initially sponsored by Bristol-Myers Squibb in 2006 [43]. The first trial was a multicenter, single-arm trial of previously treated, unresectable stage III or IV melanoma patients treated with ipilimumab monotherapy at 10 mg/kg every 3 weeks for four cycles, followed by repeat dosing every 3 months. The primary endpoint of best overall response rate (BORR), defined as the proportion of patients with compete response (CR) or partial response (PR). The study defined response rate by the traditional modified World Health Organization (WHO) criteria. The trial reported a 5.8 % BORR with a higher rate of response (27 %) when stable disease was included (defined as disease control rate, DCR) [72].
The second phase II ipilimumab monotherapy trial evaluated different dosing rates in randomized, double-blinded fashion. Again, pretreated unresectable stage III or IV melanoma patients were enrolled and dosed at 0.3, 3, or 10 mg/kg every 3 weeks for four cycles followed by maintenance therapy every 3 months at the same dose. As in the previous study, the primary endpoint was BORR by WHO criteria. This trial noted a dose-dependent relationship in both efficacy and irAEs. The trial found a BORR of 0 % for 0.3 mg/kg dosing, 4.2 % for 3 mg/kg dosing, and 11.1 % for 10 mg/kg dosing. The rates of serious (≥grade 3) irAEs were 0, 7, and 25 % for the dosing groups, respectively. This trial helped establish that 3 mg/kg was the minimum biologically active dose [73].
The third phase II trial examined the effect of inclusion of budesonide specifically to prevent treatment-induced diarrhea, along with ipilimumab. The trial included both treatment-naïve and pretreated patients with unresectable stage III and stage IV melanoma. Ipilimumab was given open-label at 10 mg/kg every 3 weeks for four doses followed by maintenance therapy every 3 months. Patients were randomized and blinded to receive daily budesonide or placebo. Budesonide treatment did not affect the efficacy of ipilimumab which demonstrated a similar BORR in both arms and also comparable to the previous phase II trials. The BORR were 12.1 % (ipilimumab with budesonide) and 15.8 % (ipilimumab with placebo), with similar 1- and 2-year overall survival (OS). Prophylactic budesonide did not affect the rates of ≥ grade 2 diarrhea or other irAEs [74]. A retrospective evaluation of the data from this trial determined that treatment-naïve patients had significantly longer survival (median OS 30.5 months) compared to pretreated patients (median OS 13.6 months) [75].
An additional phase II trial, reported after ipilimumab was FDA-approved, looked specifically at melanoma patients with brain metastases, a group that had been excluded from most of the previously performed studies. The patients were divided into groups depending on whether their brain lesions were symptomatic or not. Patients were given ipilimumab at 10 mg/kg every 3 weeks with maintenance therapy. The trial found both groups responded at a similar low rate to previous trials with comparable toxicity. Patients with small, asymptomatic brain metastases generally responded better than larger or symptomatic lesions [76].
Phase III Trial of Ipilimumab Monotherapy in Melanoma
The first phase III study of ipilimumab, sponsored by Bristol-Myers Squibb, began enrolling patients in September 2004. The trial enrolled 676 HLA-A*0201+ patients with pretreated, unresectable stage III or IV melanoma. The patients were randomized 3:1:1 to receive either ipilimumab with gp100 peptide vaccine, ipilimumab alone, or gp100 alone. The gp100 peptide had demonstrated effectiveness in previous phase II trials in melanoma, particularly when combined with ipilimumab [71, 77–79]. Ipilimumab was dosed at 3 mg/kg every 3 weeks for four doses. Patients were not routinely offered maintenance therapy; however, those who progressed after responding to therapy or who had stable disease after 12 weeks were allowed “reinduction” therapy. The primary endpoint of the trial was OS. The trial demonstrated an OS benefit in all patients who received ipilimumab (median OS: 10.0 months for ipilimumab with gp100, 10.0 months for ipilimumab alone, and 6.4 months for gp100 alone; p < 0.003). There was no difference in survival in patients who received ipilimumab with gp100 and those who received ipilimumab alone. There were four cases of complete responses and multiple cases of long-term disease control in patients who received ipilimumab. Approximately, 60 % of patients treated with ipilimumab experienced some irAE, with the rates of serious irAEs (≥grade 3) of 10–15 % in the ipilimumab groups [80]. Of the 31 patients who met criteria for and received “reinduction” therapy (progression after complete or partial response or stable disease), 19 % achieved a complete or partial response and 68 % achieved disease control with similar toxicity to the original induction therapy [81]. Based on this study, ipilimumab achieved FDA approval at a dose of 3.0 mg/kg to treat unresectable stage III and stage IV melanoma.
When ipilimumab was approved for therapy, it generated considerable interest because it represented a therapeutic success for nonspecific immunostimulation, a new modality in cancer treatment. In addition to this, it raised hope for future successes for cancer immunotherapy, particularly coming on the heels of the FDA approval of another cancer immunotherapy, sipuleucel T (Provenge; Dendreon, Seattle, WA), the first therapeutic cellular immunotherapy to prove effective in phase III trials [5, 6]. It gave hope to clinicians treating and patients with metastatic melanoma, as this was the first therapy to show an overall survival benefit in a randomized, phase III trial for metastatic melanoma [82]. Significant questions remain and are currently under evaluation regarding the treatment of melanoma with ipilimumab. As discussed previously, a randomized, double-blinded phase II trial comparing the dosing of ipilimumab demonstrated the superiority of 10 mg/kg dosing over 3 mg/kg dosing (used in the phase III trial and currently approved) in pretreated patients [73]. This data was not available at the initiation of the phase III trial. A phase III trial comparing the two dosing levels in both pretreated and untreated metastatic melanoma patients is currently underway (NCT01515189).
An additional question raised by the previous trials is the duration of treatment. Many of the previous phase II trials included maintenance dosing every 3 months after completion of the “induction” phase [72–74, 83]. The phase III trial of ipilimumab monotherapy applied a somewhat different approach, using “reinduction” therapy, in which the patients were redosed every 3 weeks for four doses if they had evidence of progression after initial response to treatment. Both long-term dosing schedules appear to be well tolerated. It remains to be seen if one is clearly superior.
Since ipilimumab has been approved, another drug, vemurafenib, a BRAF inhibitor, has proven effective and been FDA approved in patients with BRAF-V600E mutated (present in about 50 % of patients) metastatic melanoma [84]. The presence of a BRAF-V600E mutation does not appear to affect responsiveness to ipilimumab [85]. The optimal timing and sequence of these drugs in patients eligible for both has yet to be determined. Finally, the effectiveness of ipilimumab in patients with resectable high-risk stage III and stage IV melanoma is currently under investigation in two clinical trials (NCT01274338, NCT01274338). Other areas of active investigation in ipilimumab monotherapy include identification of the subset of patients who benefit most from CTLA-4 blockade and biomarkers predicting response to therapy.
6.2.3.2 Tremelimumab
Tremelimumab (formerly CP-675, 206, ticilimumab, previously licensed to Pfizer, New York, NY, now licensed to AstraZeneca, London, UK) is another humanized anti-CTLA-4 mAb that has been evaluated in human clinical trials [18, 86]. Tremelimumab is an IgG2 antibody that, similar to ipilimumab, blocks the binding site of CTLA-4. It has a longer half-life of approximately 22 days compared to 12–14 days for ipilimumab [86]. In vitro testing of tremelimumab revealed enhanced T-cell activation, demonstrated by increased cytokine production. Based on this, as well as initial experience with ipilimumab, the drug proceeded with human trials.
The first dose escalation phase I trial of tremelimumab enrolled metastatic melanoma (n = 34), renal cell carcinoma (n = 4), and colon cancer patients (n = 1). The trial did note dose-limiting autoimmune toxicity, but determined that the drug was tolerated up to 15 mg/kg in a single dose. The trial also noted complete or partial response in 4 of the 29 patients with measurable melanoma [87].
A phase I/II trial further evaluated dosing in metastatic melanoma patients and recommended dosing at 15 mg/kg every 3 months for further study given equivalent efficacy and better safety to more frequent dosing [88]. A subsequent single-arm, phase II trial of tremelimumab was conducted in 251 patients with relapsed or refractory metastatic melanoma. Patients were treated with tremelimumab at 15 mg/kg every 90 days (as recommended in the previous trial) for four doses and allowed up to four additional doses in patients with a tumor response or stable disease. The trial revealed an objective response rate of 6.6 %. The trial reported an overall OS of 10.0 months, which is comparable with what was found in the previously described phase III trial of ipilimumab in similar patients. Serious adverse events (≥grade 3) were seen in 21 % of patients [89].
The phase III trial of tremelimumab monotherapy in treatment-naïve unresectable stage III or stage IV melanoma began enrolling in March 2006. Patients were randomized to receive tremelimumab at 15 mg/kg every 90 days until symptomatic disease progression or standard-of-care chemotherapy (temozolomide or dacarbazine) for 12 weeks or until disease progression. The primary end-point was OS. The trial was terminated by the data safety monitoring board at the second interim analysis (after two-thirds of planned events had occurred) because the test statistic crossed the prespecified futility boundary [90]. Survival follow-up continued after the trial was stopped. At final analysis, the median overall survival was 12.6 months in the tremelimumab arm compared to 10.7 months in the chemotherapy arm (p = 0.127). Objective response rates were similar in both arms (10.7 % vs. 9.8 %, respectively). Grade 3 or 4 adverse events occurred in 52 % of tremelimumab patients compared to 37 % of chemotherapy patients [91]. More recent work has suggested that the lack of tremelimumab efficacy may stem from the fact that it is an IgG2 isotype mAb, thus less able to produce reduction in intratumoral Tregs than ipilimumab, an IgG1 mAb [26]. Despite its lack of proven effect in this trial, tremelimumab remains under active investigation in other patient populations (discussed further below).
6.2.4 Toxicity
As previously described, CTLA-4 blocking antibodies can lead to unique, immunologic toxicities termed “immune-related adverse events” (irAEs) through nonspecific activation of the immune system. While the majority of these are minor and manageable, they occur relatively frequently, particularly at higher doses and can be severe. In the first phase III trial of ipilimumab, with treatment at 3 mg/kg, 14 patients (2.1 %) receiving ipilimumab died from causes deemed treatment related, 7 of the deaths were from irAEs [80]. In a pooled analysis of 325 patients treated with ipilimumab at 10 mg/kg every 3 weeks for four doses, 72.3 % experienced irAEs and 25.2 % were ≥ grade 3 [92]. In the phase III trial combining ipilimumab with dacarbazine for treatment-naïve melanoma, 56.3 % of patients in the combination arm experienced grade 3 or 4 adverse events. The most frequent irAEs are of the skin, gastrointestinal tract, liver, and endocrine system. These adverse events tend to occur at predictable times after receiving CTLA-4 blocking antibodies [92].
Skin toxicity is the most frequent irAE in some series, with roughly half of the patients receiving ipilimumab experiencing some form of rash. The rashes can typically be managed with symptom control and topical medication until they become more severe when systemic steroids and/or withholding or discontinuing treatment may be necessary. There are rare reported cases of toxic epidermal necrolysis that have been fatal [74].
Diarrhea is another frequent adverse event seen in CTLA-4 blockade treatment, occurring in between 32.8 and 51 % of patients in phase III trials of ipilimumab and tremelimumab [80, 91, 93]. Severe diarrhea, colitis, and perforation are less common but can occur. Like skin toxicity, initial management is symptomatic. A high degree of suspicion for colitis with a low threshold for endoscopic evaluation is necessary for more severe (≥grade 2) diarrhea. The diagnosis of colitis or grade 3 or higher diarrhea necessitates more aggressive treatment with fluid replacement, systemic steroids, and treatment cessation. Infliximab treatment has been effective for severe colitis. A high index of suspicion for perforation with involvement of gastroenterology and surgery is also warranted in these cases [74].
Hepatotoxicity is seen less frequently (3–9 %) with CTLA-4 blocking antibodies but can be severe. In general, liver function tests should be followed during treatment and ≥ grade 3 hepatotoxicity requires systemic treatment with systemic steroids and occasionally mycophenolate mofetil along with drug cessation [92].
Endocrine toxicities consist of hypophysitis and, less frequently, autoimmune thyroid dysfunction and adrenal insufficiency. Hypophysitis appears to occur in less than 5 % of cases but typically has permanent sequelae and can lead to life-threatening adrenal insufficiency if not properly recognized and managed. Suspicion for hypophysitis should lead to pituitary MRI and laboratory testing. Treatment consists of systemic steroids and withholding CTLA-4 blocking treatment. Monitoring of serum chemistries and thyroid function panels is recommended with ipilimumab treatment [94].
Other less frequent irAEs seen with CTLA-4 blocking therapies include episcleritis, uveitis, pancreatitis, neuropathies, and lymphadenopathy. Screening for a history of autoimmune disease and consideration of risk factors and expected benefits is recommended given the potential for serious toxicity with CTLA-4 blocking antibodies. National Comprehensive Cancer Network (NCCN) guidelines recommend participation in a risk evaluation and mitigation strategy (REMS) program when using ipilimumab [95].
Interestingly, multiple phase I and II trials of ipilimumab have noted a higher rate of clinical response in patients with irAEs and, in particular, grade 3 and 4 irAEs [71, 74, 78, 79, 96–99]. A similar correlation was not addressed in the phase III trials of CTLA-4 blockade antibodies, and further evaluation may help clarify this as well as the underlying mechanisms.
6.2.5 Immune-Related Response Criteria
Initial WHO response criteria and later RECIST criteria, which have undergone many revisions over the years, were developed to identify and standardize definitions of tumors responsive to cytotoxic therapy and not as a surrogate for survival [100]. They have been used in early phase clinical trials as a surrogate for response to therapy. The use of these criteria assumes that tumors will shrink or stabilize at the outset of therapy. Tumor growth or the appearance of new metastases constitute progressive disease and, therefore, lack of response. In immunotherapy trials, including those evaluating ipilimumab, it has been shown that tumors often progress or remain stable before responding, therefore making RECIST criteria less helpful in predicting treatment response. Based on these observations, new immune-related response criteria (irRC) were proposed (Table 6.1). The new criteria do not necessarily consider the appearance of new lesions or growth of isolated lesions as progressive disease but, instead, consider overall tumor burden. Based on retrospective observations of 487 metastatic melanoma patients in three phase II trials of ipilimumab at 10 mg/kg dosing, 9.7 % of treated patients initially classified as progressive disease under WHO criteria later had evidence of response to therapy. In retrospective reclassification by irRC, response to therapy appears to correlate better with overall survival than WHO criteria [101]. Immune-related response criteria have been used alongside WHO criteria in multiple ipilimumab trials since it was first introduced [72, 102]. Further prospective validation will be needed to determine to what degree it correlates with overall survival.
Table 6.1
Comparison of World Health Organization (WHO) and immune-related response criteria (irRC) for tumor response [101]
World Health Organization (WHO) | Immune-related response criteria (irRC) | |
|---|---|---|
CR | Disappearance of all lesions in two observations at least 4 weeks apart | Disappearance of all lesions in two observations at least 4 weeks apart |
PR | ≥50 % decrease in SPD of all index lesions in the absence of progression of nonindex lesions or new lesions in two observations at least 2 weeks apart | ≥50 % decrease in total tumor burden in two observations at least 4 weeks apart |
SD | <50 % decrease compared to baseline and <25 % increase compared to nadir measurements of the SPD of index lesions, in the absence of progression of nonindex lesions or new lesions | <50 decrease compared to baseline and <25 % increase compared to nadir |
PD | ≥25 % increase in SPD compared with nadir or progressions of nonindex lesions or appearance of new lesions | ≥25 % increase in tumor burden compared to nadir in two observations at least 4 weeks apart |
6.2.6 CTLA-4 Blockade in Cancers Other than Cutaneous Melanoma
As previously discussed, melanoma was a logical first target for CTLA-4 blockade, given the evidence that it is an immunogenic tumor [103]. Given the success found in treatment of cutaneous melanoma, the use of CTLA-4 blockade alone or in combination with other therapies to treat other tumor types is an active area of investigation.
6.2.6.1 Uveal Melanoma
Uveal melanoma is a rare cancer that, like cutaneous melanoma, shares melanocytes as the cell or origin but has different pathogenesis and clinical behavior. Similar to melanoma, it has a very poor prognosis when it has metastasized (typically to the liver) and is resistant to systemic chemotherapy [104]. Uveal melanoma was excluded from most previous studies of ipilimumab in melanoma. A retrospective look at 14 patients with metastatic uveal melanoma treated with ipilimumab at 10 mg/kg from seven European centers’ compassionate use program revealed a 29 % rate of partial response or stable disease with response behavior similar to those seen in cutaneous melanoma [105]. Phase I/II trials looking at ipilimumab in the adjuvant setting for high-risk uveal melanoma after completion of standard treatment and for the treatment of metastatic uveal melanoma are underway (NCT01585194).
6.2.6.2 Prostate Cancer
Castrate-resistant prostate cancer (CRPC) has limited treatment options available; however, it has proven susceptible to immunotherapy. As previously discussed, one of the original phase I trials of ipilimumab was conducted in CRPC patients and demonstrated a transient decline in the PSA in a number of patients [106]. An additional phase I dose escalation trial was conducted using tremelimumab in combination with androgen deprivation (with bicalutamide) in patients with PSA-recurrent prostate cancer after primary surgery or radiation. Out of 11 patients, 3 were noted to have late prolongation in their PSA doubling time, and toxicities were generally mild [107].
There have been five phase II trials of ipilimumab in prostate cancer with androgen deprivation therapy, radiation, chemotherapy, or immunotherapy and are described further in subsequent sections [108]. Two phase II trials have included an ipilimumab monotherapy arm that produced PSA declines of >50 % in a minority (13–25 %) of patients [109, 110].
There are currently nine ongoing phase II and two phase III trials involving ipilimumab in prostate cancer, most of which involve combination therapy [108, 111]. In addition, there is a randomized phase III trial of ipilimumab monotherapy in metastatic asymptomatic or minimally symptomatic CRPR currently underway and scheduled to have results in November 2015 (NCT01057810). The results of the phase III trials will be interesting, as they will examine overall survival. This is particularly relevant in light of the fact that sipuleucel-T, a cancer vaccine approved for treatment of metastatic CRPC, proved to be effective at improving overall survival without significantly decreasing PSA levels, which has been the primary endpoint for the phase II trials of CTLA-4 blocking therapy [5].
In addition to monotherapy, CTLA-4 blockade has been tried in combination with androgen deprivation therapy. Androgen deprivation is the first-line therapy for recurrent or metastatic prostate cancer and is associated with prostatic tissue apoptosis and lymphocytic infiltration. There is also evidence that androgen deprivation may stimulate thymopoiesis and specifically the production of naïve T-cells which may enhance antitumor response [112]. CTLA-4 blockade may augment the natural immune response elicited by this treatment. A phase I trial of tremelimumab in combination with androgen deprivation (with bicalutamide) was conducted in patients with PSA-recurrent prostate cancer as discussed earlier [107]. A phase II trial was conducted in which 108 patients with advanced prostate cancer were randomized to receive ipilimumab, given as a single dose of 3 mg/kg, in combination with androgen deprivation or androgen deprivation alone. Fifty-five percent of patients with combination therapy had a PSA reduction >50 % compared to 38 % in the arm undergoing androgen deprivation only [113]. Additional trials of this combination are currently underway (NCT01377389, NCT01498978).
6.2.6.3 Breast Cancer
Similar to the aforementioned trial in prostate cancer, tremelimumab has been used in combination with exemestane, an aromatase inhibitor, in patients with advanced breast cancer. Twenty-six patients with advanced estrogen and/or progesterone receptor-positive breast cancer, all of whom had progressed on previous hormonal therapy, were treated with various doses of tremelimumab in combination with exemestane as part of a phase I dose escalation trial. The maximum tolerated dose was determined to be 6 mg/kg dosed every 90 days. This is a lower dose than used in previous trials, likely because dose-limiting toxicity was defined to include some grade 2 toxicities if they were immune related. The best response to therapy was stable disease for at least 12 weeks, which was noted in 42 % of patients. There was a correlation between patients with disease stability and peripheral lymphocytes expressing ICOS (inducible costimulator), a member of the CD28-superfamily of costimulatory molecules expressed on activated T-cells [114].
6.2.6.4 Renal Cell Carcinoma
A phase II trial of ipilimumab monotherapy was conducted in patients with metastatic renal cell carcinoma, a tumor thought to be immunogenic given rare observed cases of spontaneous regression and its observed response to cytokine therapy. Ipilimumab was dosed at 3 mg/kg initially followed by either 1 mg/kg or 3 mg/kg dosing every 3 weeks. Five out of 40 patients had partial response by RECIST criteria, including patients who had not previously responded to IL-2 therapy. As in other trials, they reported a correlation between response to ipilimumab therapy and autoimmune toxicities [97].
6.2.6.5 Gastrointestinal Cancers
There has been one phase II trial of tremelimumab as second-line therapy for metastatic gastric or esophageal carcinoma. Patients were given tremelimumab at 15 mg/kg every 3 months and monitored for response by RECIST criteria. One of 18 patients had a partial response and remained well on treatment for 33 months, while 4 others had stable disease. There was one treatment-related death from autoimmune colitis [115].
Metastatic and unresectable pancreatic adenocarcinoma is another disease process with few effective treatment options. A phase II trial of ipilimumab monotherapy was conducted in 27 pancreatic cancer patients, with treatment given at 3 mg/kg every 3 weeks for four doses. No patients responded by RECIST criteria, although there was one late response observed [116].
A phase II trial of tremelimumab monotherapy was conducted in patients with heavily pretreated metastatic colon cancer. Tremelimumab was given at 15 mg/kg every 3 months until disease progression. Of the 45 evaluable patients, only 44 received a single dose secondary to disease progression (n = 43) or discontinuation (n = 1). One patient had a partial response and received therapy for 15 months [117].
6.2.6.6 Hepatocellular Carcinoma
CTLA-4 blockade showed a particularly promising effect on patients with advanced hepatocellular carcinoma and chronic hepatitis C infection, another cancer with limited treatment options and a tumor that does have a history of both spontaneous regression and response to immunotherapy [118]. Tremelimumab was given at 15 mg/kg every 3 months to 20 patients with advanced hepatocellular carcinoma, many of whom were Child-Pugh class B cirrhotics (class C patients were excluded) and were not amenable to surgery and chronic hepatitis C infection; treatment was given until disease progression or severe toxicity. RECIST criteria PR were seen in 17.6 % of patients with a reported DCR in 76 %. Interestingly, along with the impressive tumor responses, a significant drop in hepatitis C viral load coupled with enhanced specific antiviral immunity was observed, raising the question as to whether CTLA-4 blockade may be beneficial in virus-associated malignancies [119].
6.2.7 CTLA-4 Blockade as Combination Therapy
While CTLA-4 blockade, specifically ipilimumab, has found success as monotherapy in metastatic melanoma, and more trials are underway to test its effectiveness in a variety of malignancies and different clinical scenarios, its greatest potential may lie in combining it with other antineoplastic agents. The hope is that by combining CTLA-4 blocking therapy with other antineoplastic therapies that carry different toxicity profiles a synergistic effect of the agents will be achieved. Recognizing these issues, researchers have been actively pursuing combination therapy with CTLA-4 blockade since its inception. The primary areas of research focus on combining CTLA-4 blockade with chemotherapy, radiation, surgery, and other immunotherapy.
6.2.7.1 CTLA-4 Blockade and Chemotherapy
Given the known immunosuppressive effects of most chemotherapeutic agents, it has been thought that combining chemotherapy with immunotherapy would be unsuccessful. However, there is increasing evidence for a possible synergistic role between the two modalities. The immune system appears to play an important role in antitumor activity of chemotherapy, an effect which may be further augmented by immune checkpoint blockade [123, 124]. In murine models of mesothelioma, CTLA-4 blockade given between cycles of chemotherapy has been demonstrated to increase tumor-infiltrating lymphocytes and inflammatory cytokines and inhibit cancer cell repopulation [125]. Additionally, chemotherapy, when given appropriately, may enhance the effect of specific immunotherapy [126]. Evidence from clinical trials reveals that combining chemotherapy with cancer vaccination can be more effective than either therapy alone [127–129]. The mechanisms by which chemotherapy may increase anticancer immunity include reduction of immunosuppressive influences by decreasing tumor mass, inducing the expression of TAAs on the cell surface, exposing the immune system to TAAs through cell death, and “resetting” the immune posture through depletion of inhibitory cell populations (i.e., Tregs and myeloid-derived suppressor cells) [123]. Indeed, there is growing evidence that the success of certain chemotherapy regimens is dependent on the drug’s ability to cause immunogenic cell death of tumors, where TAAs are presented in the appropriate context to elicit a broader immune response [130]. While this is a promising area for future development, clearly the timing of drug administration, chemotherapeutic regimen used, and dosing are integrally important to successful application. Highly dosed cytotoxic treatment has the potential to quash a developing therapeutic immune response. Optimizing these factors will be necessary in future trials of combining checkpoint blockade with chemotherapy.
Clinical trials have been performed combining chemotherapy with CTLA-4 blockade. A randomized phase II trial testing the combination of chemotherapy with ipilimumab was conducted in patients with treatment-naïve metastatic melanoma. Seventy-two patients with unresectable, metastatic melanoma were randomized to receive ipilimumab at 3 mg/kg every 4 weeks for four doses with dacarbazine compared to ipilimumab monotherapy. The trial demonstrated an increased objective response rate (14.3 % vs. 5.4 %, by RECIST criteria) and increased median OS (14.3 vs. 11.4 months) for the combination therapy group, although neither reached statistical significance due to the smaller number of patients. Toxicity was higher in the combination group, including 17.1 % ≥ grade 3 irAEs compared to 7.7 % in the monotherapy arm [131].
Based on these results, the concept was tested in a randomized phase III trial evaluating ipilimumab with dacarbazine vs. dacarbazine alone [73]. Additionally, based on the results of the phase II ipilimumab monotherapy trial that showed a benefit of higher dosing, 10 mg/kg of ipilimumab was used in combination with dacarbazine. Five hundred two patients were enrolled and randomized 1:1 to receive ipilimumab plus dacarbazine every 3 weeks for four doses followed by dacarbazine every 3 weeks until week 22 or placebo plus dacarbazine at the same schedule. Patients with stable disease or RECIST criteria objective responses were able to receive maintenance ipilimumab or placebo every 12 weeks. Of note, based on emerging consensus from previous work with CTLA-4 blockade and other immunotherapy, the primary endpoint was changed, with FDA approval, from progression-free survival to OS prior to unblinding of the treatment groups or data analysis [101, 132]. Ultimately, the trial showed that patients who received the combination of ipilimumab with dacarbazine survived longer (11.2 months) compared to dacarbazine alone (9.2 months, p < 0.001). The difference became more pronounced with time, as the combination arm had 20.8 % of patients alive at 3 years compared to 12.2 % in the chemotherapy only arm. Toxicities were greater in the combination arm and also greater than in many of the previous ipilimumab studies (56 % ≥ grade 3), likely secondary to the higher dose (10 mg/kg) of ipilimumab used as well as the addition of chemotherapy. Interestingly, the toxicity profile was different. There were lower rates of gastrointestinal toxicities, such as diarrhea and colitis, and endocrine toxicity but a higher rate of hepatic toxicity compared with previous ipilimumab trials. No treatment-related death was reported [93]. Differences may reflect the effect of the combination therapy; however, clinician’s experience managing the drug may have affected the outcome as well. Based on the results of this study, the combination of ipilimumab and dacarbazine is approved as the first-line therapy for unresectable melanoma.
However, the potential for unanticipated toxicity exists with combining CTLA-4 blockade, particularly with other targeted therapies. Initial results from a phase I study of combination therapy with both ipilimumab (dosed at 3 mg/kg) and vemurafenib, a BRAF inhibitor approved for treatment of BRAF-V600E mutated melanoma, demonstrated an unacceptably high level of hepatotoxicity, leading to early termination of the trial [133].
Additional trials of combination chemotherapy and ipilimumab were conducted in patients with advanced non-small-cell lung cancer (NSCLC) and small cell lung cancer (SCLC). Advanced-stage NSCLC carries a poor prognosis with a median survival of 8–12 months despite first-line chemotherapy [124, 134]. In a phase II trial, 204 patients with stage IIIB or IV NSCLC were enrolled in a randomized, double-blind trial of ipilimumab plus chemotherapy (paclitaxel and carboplatin) given concurrently, ipilimumab plus chemotherapy given phased with two doses of chemotherapy given prior to starting ipilimumab and chemotherapy given together, or placebo plus chemotherapy. Ipilimumab was dosed at 10 mg/kg every 3 weeks for up to 18 weeks with the option for maintenance therapy (or maintenance placebo) every 12 weeks. The primary endpoint was immune-related progression-free survival (irPFS). The concept of immune-response criteria for immunotherapy in cancer (different from classic World Health Organization RECIST criteria) came from observations with ipilimumab and other immunotherapies (discussed further below) [101]. The trial showed improved irPFS with phased ipilimumab and chemotherapy (median: 5.7 months, HR: 0.72, p = 0.05), while concurrent ipilimumab and chemotherapy did not reach statistical significance (median: 5.5 months, HR: 081, p = 0.13) compared to the control regimen (median 4.6 months). Improvement was also noted in PFS by WHO criteria (p = 0.02), and an improvement in OS by 3.9 months (p = 0.23) was observed for phased ipilimumab over chemotherapy alone. Overall toxicity was similar across the treatment arms; however, there was more severe toxicity (grade ≥3) in the combination arms. A phase III trial is being conducted using phased ipilimumab and chemotherapy in patients with squamous NSCLC, the group that derived the greatest benefit in subset analyses [102] (NCT01285609).
A similar phase II trial was conducted in patients with extensive disease small-cell lung cancer (ED-SCLC). Chemotherapy remains the first line and only effective therapy in this disease process with a median overall survival of 8–11 months [135]. Eligible patients (n = 130) were randomized to receive concurrent therapy with ipilimumab and chemotherapy (paclitaxel and carboplatin), the phased combination, or placebo with chemotherapy. In this trial, again the phased combination of ipilimumab and chemotherapy was superior with an improvement in irPFS (median: 6.4 months, p = 0.03), while concurrent therapy did not improve irPFS (median: 5.7 months, p = 0.11), compared to the control arm (median: 5.3 months). There was no significant difference in mWHO PFS or OS. The phased combination of ipilimumab and paclitaxel/carboplatin is currently being tested in a phase III trial with an anticipated enrollment of 912 patients (NCT01450761).
The combination of ipilimumab has been further studied in a phase II trial in prostate cancer. Forty-three patients with CRPC were randomized to receive either ipilimumab monotherapy at 3 mg/kg every 3 weeks for four doses or ipilimumab (dosed the same) with a single dose of docetaxel at the start of therapy. The number of responses to therapy was small with three patients having a decrease of >50 % in each arm [109]. However, this study may be limited by underdosing of both the ipilimumab and docetaxel, concurrent (instead of phased) administration of the two drugs, as well as the small number of patients tested.
The combination of tremelimumab and sunitinib, an oral small-molecule tyrosine kinase inhibitor, was tested in a phase I dose escalation trial in patients with metastatic renal cell carcinoma. Unexpectedly, the trial demonstrated a high (4/28 patients) rate of sudden onset grade 3 renal failure in addition to other toxicity associated with CTLA-4 blockade. Further testing of this combination at doses of tremelimumab >6 mg/kg with sunitinib was not recommended by the study authors [136].
6.2.7.2 CTLA-4 Blockade and Radiation
Much like chemotherapy, there is evidence that the local and systemic effects of radiation therapy can increase the effectiveness of immunotherapy, in general, and CTLA-4 blockade, specifically. Radiation therapy damages tumor cells that are in the path of the focused energy, which, like chemotherapy, can result in cell death and antigen cross-presentation, leading to an effective, targeted immune response toward remaining tumor cells [137]. Radiation-induced cell damage may lead to several cellular changes that promote effective presentation of TAAs such as the release of high mobility box group 1 (HMBG1), which signals migration of immune cells to the tumor microenvironment, and upregulation of MHC I complexes, Fas, and ICAM-1, all of which increase susceptibility to T-cell-mediated death [137–140]. Additionally, localized radiation does not typically produce the same level of lymphodepletion and immunosuppression associated with high-dose chemotherapy. As with chemotherapy, reduction in the mass of a viable tumor may help decrease cancer-related immunosuppression. All of these factors make the combination of radiation with immunotherapy appealing [141]. The concept of combining radiation with immune checkpoint blockade is particularly attractive. Unlike more specific, directed immunotherapy (cancer vaccines), CTLA-4 blockade helps overcome cancer immunosuppression, but ultimately relies on the body’s preexisting immunity toward a neoplasm. Radiation, by damaging cancer cells and releasing a wide array of TAAs in an inflammatory context, especially with immunosuppression checked, may allow the immune system to mount a response that is appropriate both for the individual and the tumor.
There is considerable preclinical data that supports the combination of CTLA-4 blockade and radiation. In one study, a mouse model of poorly immunogenic mammary carcinoma, 4T1, was treated with control IgG, CTLA-4 blocking IgG (9H10), radiation therapy, or a combination of 9H10 IgG and radiation. CTLA-4 blockade alone did not affect tumor growth or mouse survival. Radiation therapy slowed tumor growth but did not affect survival. The combination of CTLA-4 blockade and radiation therapy inhibited metastases and increased survival compared to the control [141]. Subsequent studies in this model revealed that treatment with the combination in mice deficient in invariant natural killer (NK) T-cell lymphocytes led to an even more effective response with some mice becoming disease-free and resistant to tumor rechallenge, highlighting the important role for this cell type in regulation of cancer immune responses [56]. Finally, an additional study in TSA mouse mammary carcinoma and MCA38 mouse colon carcinoma models again demonstrated the effectiveness of combining radiation and CTLA-4 blocking antibody; moreover, they showed that the use of a fractionated radiation schedule (but not single dose radiation) along with CTLA-4 blockade could significantly inhibit tumor foci out of the radiation field, a phenomenon known as the abscopal effect [55].
The abscopal effect refers to the regression of tumors in remote areas following localized radiation of tumors. These phenomena have been documented in melanoma, renal cell carcinoma, and lymphoma [142–144]. More recently, several cases of this occurrence have been documented in patients receiving ipilimumab. In one notable case, a patient with recurrent melanoma with paraspinal, right hilar lymphadenopathy, and splenic metastases was enrolled in an ipilimumab monotherapy trial in September 2009. She received treatment at 10 mg/kg dosing per protocol with slow progression of her disease over the subsequent 15 months. In December 2010, she received directed, external beam radiation to her symptomatic paraspinal lesion followed by an additional dose of ipilimumab in February 2011. Surprisingly, follow-up imaging revealed significant regression of metastatic lesions outside the radiation field, which remained stable at minimal disease for at least 10 months after her radiation treatment. Along with this clinical effect, the patient was noted to have a marked increase in peripheral antibodies to the tumor antigen NY-ESO-1, an increase in ICOShigh T-cells, and a decrease in myeloid derived suppressor cells [145]. Similar cases of abscopal regression of metastatic melanoma in patients on ipilimumab have since been reported [146].
A phase I/II examined the effects of ipilimumab with radiation therapy (RT) in patients with metastatic CRPC. Patients were treated with dose escalation ipilimumab monotherapy (3, 5, or 10 mg/kg) or ipilimumab (3 mg/kg or 10 mg/kg) with external beam RT, although the trials were not designed to directly compare the two arms. Ipilimumab was given every 3 weeks for a total of 4 weeks [110]. An overall of 71 patients were treated; 33 patients were treated in the dose escalation phase and the 10 mg/kg arm was expanded to a total of 50 patients. At the 10 mg/kg dosing level, 16 were given ipilimumab monotherapy and 34 received ipilimumab with radiation. In the 10 mg/kg dosing group, there were four (25 %) PSA declines >50 % in the ipilimumab monotherapy arm and four (12 %) PSA declines >50 % in the ipilimumab with radiation group; however a higher proportion of patients in the monotherapy group were chemotherapy naïve [110]. A phase III trial examining radiation with ipilimumab compared to radiation alone in advanced CRPC is currently underway (NCT00861614). Additional phase I/II and II trials evaluating the effect of ipilimumab with and without radiation in multiple cancer types are currently underway (NCT01689974, NCT01769222, NCT01449279).
6.3 Programmed Death 1 (PD-1) Pathway
6.3.1 Function
Programmed death 1 (PD-1) is a more recently discovered immune checkpoint receptor that has generated considerable excitement based on favorable preclinical profiling and initial clinical results. PD-1 was first discovered in 1992 by subtractive mRNA hybridization in an attempt to identify genes involved in programmed cell death [147]. Its protein structure was deduced based on the mRNA sequence obtained; however, its function remained unclear until PD1−/− knockout mice were noted to develop lupus-like autoimmune disease [148]. At that time, it was correctly suspected that PD-1 played a role in inducing peripheral tolerance.
Since its discovery, the function and significance of PD-1 have become more clear [149]. Like CTLA-4, PD-1 is a transmembrane protein expressed on effector immune cells [150]. Also like CTLA-4, expression of PD-1 is inducibly expressed with lymphocyte activation, although it is expressed more broadly than CTLA-4 as it is also found on activated B-lymphocytes and NK cells [151–153]. PD-1 is bound principally by programmed death ligand 1 (PD-L1, B7-H1) but also, to a lesser degree, by programmed death ligand 2 (PD-L2, B7-DC) [154]. PD-L1 is constitutively expressed in certain tissues such as lung and placental macrophages [155]. Its high level of expression in the placenta has been implicated in mediating materno-fetal tolerance [156, 157]. PD-L1 expression can also be induced on a broad range of hemopoietic, endothelial, and epithelial tissues in response to pro-inflammatory cytokines, such as interferon, GM-CSF, IL-4, and IL-19 [151, 158–161]. PD-L2 expression is more limited as it is inducibly expressed on dendritic cells, macrophages, and mast cells [155].
The PD-1 receptor pathway is an important negative regulator of the immune system. PD-1 appears to play role primarily in dampening immune response in the setting of peripheral inflammation as opposed to CTLA-4, which plays a greater role in regulating T-cell activation [155]. As mentioned before, PD-1 knockout mice helped initially reveal the function of PD-1. The initial B6-PD-1−/− oncogenic mice developed varying degrees of autoimmune arthritis and glomerulonephritis by 6 months of age and exaggerated inflammatory response to infection, in contrast to CTLA-4 knockout mice who die of diffuse lymphoproliferative disease shortly after birth [14, 148, 162]. Remarkably, later PD-1−/− knockout mouse models (BALB/c- PD-1−/− and MLR- PD-1−/−) developed fatal autoimmune dilated cardiomyopathy early in life due to production of autoantibodies [163, 164]. In contrast, mice deficient in PD-L1 do not manifest autoimmunity, but can have increased accumulation of CD8+ lymphocytes in the liver and increased tissue destruction with experimental autoimmune hepatitis [165].
Ligation of PD-1, which again is found primarily on immunologic cells, counters CD28-mediated signaling through multiple mechanisms. PD-1 is phosphorylated upon ligand engagement, initiating a cascade of intracellular events [166, 167]. PD-1 signaling decreases the production of several proinflammatory cytokines such as IFN-γ, TNF-α, and IL-2 [155]. It may also serve to retard cell activation mediated via CD28 and IL-2. PD-1 ligation has also been implicated in inhibiting transcription factors and initiation of several cell death pathways [168–170]. Importantly, PD-1 and its ligands also appear to play a role in shifting lymphocyte response from activation to tolerance when exposed to antigens, an attribute that is particularly significant for cancer immunotherapy [171]. Interestingly, PD-L1 was discovered to not only function as a ligand for PD-1 but also as a receptor bound by B7-1 (CD80) capable of delivering an inhibitory signal [172]. This finding not only demonstrates the complexity of lymphocyte regulation but suggests that blockade of these molecules could result in functionally different outcomes [162].
The PD-1 and PD-L pathways have been implicated in a variety of human diseases. Higher than normal expression levels of PD-1 and single nucleotide polymorphisms of PD-1 have been implicated in multiple autoimmune diseases such as systemic lupus erythematosus, Sjogren’s disease, type 1 diabetes, and rheumatoid arthritis. As such, this pathway remains an active therapeutic target in these conditions [149]. In infectious diseases, the PD-1 and PD-L pathways play an important role in preventing unnecessary immune-mediated tissue destruction and have also been implicated in preventing the clearance of chronic viral, bacterial, and parasitic infections [155, 173].
6.3.2 PD-1 Pathway in Cancer
Just as the PD-1 pathway plays a central role in tolerance of chronic infections, it also appears to have a primary role in cancer tolerance and immune escape. PD-1 ligand expression, particularly of PD-L1 expression, has been demonstrated at various levels on a large variety of human cancer tissues. Higher expression of PD-L1 on tumor cells is associated with worse prognosis, more aggressive features, and/or resistance to immunotherapy in the large majority of cancers in which it has been characterized [174–185]. However, in some cases higher expression appears to have little influence on prognosis, as was found in NSCLC, and has even been associated with a more favorable prognosis, as found in colorectal cancer without mismatch repair (MMR) deficiency [186, 187]. CD8+ tumor infiltrating lymphocytes (CD8+ TILs) have been noted to have high levels of PD-1 expression in many cases; nonetheless, correlation between PD-L expression and prognosis is mixed [181, 186, 188, 189]. Circulating NK cells in cancer patients have been noted to express PD-1, while healthy control NK cells do not [190]. Furthermore, preclinical data demonstrates that increasing tumor expression of PD-L1 makes it less susceptible to immunotherapy, while blocking it increases its vulnerability to immune-mediated destruction [191–194].
Some of the differences observed in tumor PD-L1 expression and correlation with cancer prognosis may be due to tumor-host interaction. Two recent studies examining human melanocytic lesions and colorectal cancer found a strong positive correlation between tumor PD-L1 expression and patient survival, in contrast to the majority of tissue types previously examined. However, in addition to this, higher PD-L1 expression was associated with both increased tumor infiltrating lymphocytes and interferon gamma (INF-γ) levels or gene expression in the tumor microenvironment [187, 195]. In these cases, the higher levels of PD-L1 expression may be in response to INF-γ signaling, as observed in normal human tissue [196, 197]. Thus, upregulation of PD-L1 expression may represent an adaptive tumor response to tumor-specific immunity, termed “adaptive resistance” [195, 198]. The effective host immune response may explain the more favorable outcomes observed in these patients. Other evidence implicates different transcriptionally related oncogenic pathways in the upregulation of PD-1, which may or may not be related to external inflammatory signaling [176]. The adaptive resistance hypothesis may help further explain how tumors are able to escape immune stimulation from active immunotherapy and lead to blockade of the PD-1 pathway of particular therapeutic interest.
6.3.3 PD-1 Blockade
In preclinical studies with murine cancer models, anti-PD-1 and anti-PD-L1 blockade demonstrated antitumor effect as monotherapy and augmented the effects when given concomitant with cancer vaccination [199–204]. Similarly, ex vivo blockade of PD-1 or PD-L1 improved the ability of human lymphocytic function against tumor tissue in multiple studies [191, 205–207]. Based on the functional importance of PD-1 in cancer as well as promising preclinical therapeutic results, several blocking mAbs have proceeded to human clinical trials.
6.3.3.1 Nivolumab
Several PD-1 blocking mAbs are currently under development in human trials. Nivolumab (MDX-1106, BMS-936558, Bristol-Myers Squibb, New York, NY) is a fully humanized IgG4 mAb that binds to PD-1, blocking its binding site. It was initially tested in a phase I, dose escalation trial on 296 patients with heavily pretreated advanced melanoma (n = 104), colorectal cancer (n = 19), CRPC (n = 17), NSCLC (n = 122), and renal cell carcinoma (n = 34). Nivolumab was given at 0.3, 1, 3, or 10 mg/kg in six patient cohorts followed by expansion cohorts at 10 mg/kg. Patients were initially given a single dose and allowed additional doses if they demonstrated clinical benefit; however, the trial transitioned into a phase Ib where patients were dosed every 2 weeks and reassessed every 8 weeks. Treatment was continued for up to 96 weeks or until disease progression or complete response. Overall, treatment with nivolumab was better tolerated than treatment with CTLA-4 blocking antibodies with no maximum tolerated dose achieved. Only 14 % experienced serious (≥grade 3) drug toxicity, leading to the discontinuation of therapy in only 5 %. There were drug-related adverse events in 41 % and serious drug-related adverse events in 6 % of patients that were likely irAEs, including pneumonitis, diarrhea, colitis, hepatitis, hypophysitis, and vitiligo. Pneumonitis, which occurred in 3 % of patients, is of special interest, since it was not typically seen with CTLA-4 blocking mAbs and led to only three treatment-related deaths [208]. This toxicity may be secondary to constitutive expression of PD-L1 in alveolar macrophages.
Nivolumab treatment demonstrated substantial antitumor effect, with partial or complete responses (by RECIST criteria) observed in patients with melanoma, NSCLC, and renal cell carcinoma but not colorectal cancer or CRPC. Responses were observed across various doses at rates of 19–41 % in melanoma, 6–32 % in NSCLC, and 24–31 % in renal cell carcinoma. One patient with melanoma and one with renal cell carcinoma had complete response to treatment. Responses tended to be durable with over half of melanoma and renal cell responses lasting for greater than 1 year. In addition, disease stability and mixed response (as described in irRC) were observed in a substantial portion of patients. Further analysis of PD-L1 expression from 61 patients who had pretreatment specimens available demonstrated an objective response in 36 % of tumors expressing PD-L1 and none in PD-L1-negative tumors [208]. This data raises the possibility that PD-L1 could serve as a biomarker for response to therapy, an idea that is being actively investigated.
Nivolumab monotherapy is currently being investigated in multiple clinical trials, including phase I trials in hematologic malignancies (NCT01592370) and hepatocellular carcinoma (NCT01658878), phase II trials in renal cell carcinoma (NCT01354431), and phase III trials in NSCLC (NCT01642004, NCT01673867) and melanoma (NCT01721772). A phase I trial of nivolumab combined with ipilimumab (CTLA-4 blockade) has been published and is discussed below [209]. A phase III trial of nivolumab alone or in combination with ipilimumab in melanoma is planned (NCT01844505). As previously stated, PD-1 blockade demonstrated ability to augment cancer vaccines in preclinical studies. In addition, nivolumab is also being tested in a phase I trial combined with cancer vaccines in melanoma (NCT01176461). Nivolumab is also being tested together with chemotherapy in NSCLC (NCT01454102) and renal cell carcinoma (NCT01472081).
6.3.3.2 Other PD-1 Antibodies
A second mAb under development, MK-3475 (Merck, Whitehouse Station, NJ), is a humanized IgG4 with high-affinity binding to PD-1. MK-3475 was tested in a phase I dose escalation study in nine patients with advanced malignancy. The drug was given at 1, 3, or 10 mg/kg and redosed every 2 weeks in patients with NSCLC (n = 3), rectal cancer (n = 2), melanoma (n = 2), sarcoma, and carcinoid (n = 1, each). Initial results reveal that the drug is well tolerated with no ≥ grade 3 toxicities. A partial response was seen in one melanoma patient and stable disease was noted in several others [210]. MK-3475 is currently undergoing a large phase I trial in melanoma and NSCLC with an anticipated enrollment of 439 patients to be completed in 2015 (NCT01295827).
CT-011 (CureTech, Yavne, Israel/Teva, Petah Tikva, Israel) is a humanized IgG1 anti-PD-1 antibody that has demonstrated encouraging preclinical results. A phase I dose escalation study in 17 patients with advanced hematologic malignancies was conducted with a single dose of 0.2–6 mg/kg. The drug was well tolerated with no dose-limiting toxicities. There was evidence of clinical response with one complete response observed in a patient with follicular B-cell lymphoma and several other patients having stable disease. A phase II clinical trial of CT-011 in diffuse large B-cell lymphoma is currently underway (NCT00532259). CT-011 has shown a synergistic effect in preclinical studies when combined with cancer immunotherapy [203, 211]. CT-011 is undergoing multiple clinical trials in combination with vaccine therapy in multiple myeloma, acute myelogenous leukemia, and combined with sipuleucel-T in prostate cancer (NCT01096602, NCT01067287, NCT01420965). Phase II trials combining CT-011 with chemotherapy in pancreatic cancer, colorectal cancer, and relapsed follicular lymphoma are also ongoing [161] (NCT0131416, NCT00890305, NCT00904722).
6.3.4 PD-L1 Blockade
As previously discussed, because PD-L1 is capable of acting as both a PD-1 ligand and as an inhibitory receptor (bound by B7-1), blockade of this protein may have therapeutic effects different from PD-1 blockade. Based on these findings, development of a PD-L1 blocking antibody, MDX-1105 (BMS-936559; Bristol Myers Squibb, New York, NY), proceeded. MDX-1105 has been tested in a large phase I dose escalation clinical trial on 207 patients with advanced malignancies. Treated patients had NSCLC (n = 75), melanoma (n = 55), colorectal cancer (n = 18), renal cell carcinoma (n = 17), gastric cancer (n = 7), and breast cancer (n = 4). Patients received 0.3, 1, 3, or 10 mg/kg of the study drug every 2 weeks for up to 96 weeks or until unacceptable toxicity or disease progression.
Overall, MDX-1105 was well tolerated. A maximum tolerated dose was not achieved. Serious adverse events (≥grade 3) that were treatment related were seen in 9 % of patients. Drug-related adverse events were observed in 39 %; only 5 % were serious that were likely irAEs; common adverse events included infusion-related reactions, rash, diarrhea, and hypothyroidism, all of which were generally well tolerated.
Objective responses were seen in patients with NSCLC, melanoma, renal cell carcinoma, and ovarian cancer at doses of at least 1 mg/kg. Patients with melanoma had objective response rates of 6–29 % at various doses with three complete responses seen. Patients with NSCLC had objective responses at 3 mg/kg (8 %) and 10 mg/kg (16 %). Additionally, two (12 %) patients with renal cell carcinoma and one (6 %) with ovarian cancer demonstrated objective responses. Additional patients, including patients with colorectal cancer and pancreatic cancer, but not gastric or breast cancer, demonstrated disease stability [212]. No ongoing clinical trial has been currently registered for this drug.
Initial results of the PD-1 pathway blockade are very encouraging. The findings of objective clinical responses of up to 41 % of subgroups of patients with nivolumab and relatively high response rates in NSCLC, a disease historically resistant to immunotherapy, are unprecedented in cancer immunotherapy. Additionally, lower rates of toxicity, in particular serious irAEs, compared to CTLA-4 blockade have given hope that this pathway will yield more widely applicable and better-tolerated therapies. Much work remains and is currently in progress to bring these therapies into general clinical use. Determination of optimal dosing, duration of treatment, and the subsets of patients who benefit from treatment are all underway. As with CTLA-4 blockade, preclinical data supports a possible synergistic effect when PD-1 pathway blockade is combined with other cancer treatments such as chemotherapy, radiation, and immunotherapy; this deserves and is receiving further investigation [191, 203, 205, 213]. As these investigations move forward, one area of particular interest will be whether PD-L1 expression on tumors continues to serve as a reliable biomarker for predicted therapeutic benefit, thus increasing the ever-growing trend of more personalized, tailored treatment for individual tumors.
6.4 Combination Immunotherapy
Results from trials of CTLA-4 and PD-1 pathway blocking mAbs as monotherapy or in combination with conventional therapies are encouraging. Immune checkpoint blockade has delivered clinical responses in patients with limited or no therapeutic options remaining. However, in all of the immune checkpoint blockade trials covered, only a minority of patients have responded which is usually transient. It is true that the vast majority of the patients treated in these trials have advanced disease, are immunosuppressed, and have limited time and options remaining. Targeting earlier stage disease and combining immune checkpoint blockade with other therapies will undoubtedly yield more impressive results. However, it is naïve to think that targeting any one checkpoint will be a “silver bullet” therapy. Just as cancer, under immunologic pressure, learns to evade the immune system to become a clinically evident disease initially, as we modulate coinhibitory and costimulatory receptors, some cancers will adapt to escape through alternative pathways. Combining active immunization (cancer vaccines) with checkpoint blockade may ultimately prove effective; nonetheless, initial results have not been convincing. Other techniques under investigation, targeting multiple checkpoints simultaneously or in sequence, may limit the escape routes.
6.4.1 CTLA-4 Blockade and Vaccination
Early on in the development of CTLA-4 blocking therapy, anti-CTLA-4 antibodies were combined with cancer vaccines in preclinical models [59]. In multiple cancer animal models, tumors, which were poorly responsive to CTLA-4 blocking therapy alone or active immunotherapy alone, responded significantly better to the combination of the two [29, 52, 54, 59, 60, 63, 214–221]. These studies have helped elucidate the function and significance of the CTLA-4 receptor and have led to clinical trials in patients.
Some of the first human trials of ipilimumab used a combination of peptide vaccines from gp100, a tumor-associated antigen expressed by the majority of malignant melanomas [222]. Gp100 peptides have been shown to be immunogenic and elicit an antigen-specific T-cell response in the majority of melanoma patients [77]. One peptide, gp100:209-217(210M), when combined with IL-2 therapy, has also been shown in a randomized phase III trial to significantly increase clinical response and PFS compared to IL-2 alone in HLA*A0201+ metastatic melanoma patients [223]. Three phase I and II trials were conducted using ipilimumab combined with gp100 in unresectable melanoma patients. While these trials did not directly compare the efficacy of the addition of the peptide vaccines to ipilimumab monotherapy, they did show impressive response rates and manageable toxicity [71, 78, 79]. Based on these (and other) results, ipilimumab proceeded to the phase III trial comparing ipilimumab monotherapy, ipilimumab plus two gp100 peptides (gp100:209-217 and gp100:280-288), or the gp100 peptides alone. As previously detailed, the trial demonstrated a survival advantage for ipilimumab therapy but also showed that the addition of the peptide vaccine to ipilimumab offered no improvement over ipilimumab monotherapy [80]. It is not clear why the peptide vaccine did not prove efficacious in this setting, particularly given its proven efficacy when given with IL-2 therapy in a similar patient population. There is speculation that CTLA-4 blockade may augment CD4+ lymphocyte activity more, while gp100 peptides preferentially generate a CD8+ lymphocyte response, a hypothesis that has mixed preclinical data to support it [223]. Another proposed possibility is that the antitumor effect of ipilimumab may stem largely from its ability to deplete intratumoral Tregs, a mechanism which may not function synergistically with MHC class I peptide vaccination [26]. Certainly, there are other possibilities to explain the results; further studies will be necessary to clarify.
Additional trials on combining CTLA-4 blocking antibodies with cancer vaccines have been conducted in melanoma and prostate cancer. In melanoma, the combination of multiple tumor-associated antigen peptides (gp100, MART-1, tyrosinase) emulsified with immunoadjuvant (Montanide ISA 51) have been combined with ipilimumab in a dose escalation trial [99]. Additionally, in prostate cancer, ipilimumab has been given in phase I trials in combination with Tricom-PSA (PROSTVAC; Bavarian Nordic Immunotherapeutics, Mountain View, CA), a poxvirus-based vaccine that expresses transgenes for PSA and costimulatory molecules, and GVAX (Aduro Biotech; Berkeley, CA, USA), a GM-CSF transduced allogenic prostate cancer vaccine [96, 224]. In all these phase I trials, ipilimumab combined with cancer vaccination was found to elicit a cancer-specific immune response, a low rate of clinical response, and toxicity compared with ipilimumab monotherapy. Further trials will be necessary to prove the efficacy of these combinations and multiple other combinations which are currently under investigation (NCT01810016, NCT01302496, NCT01838200).
6.4.2 CTLA-4 Blockade and Cytokine Therapy
Another area of combined immunotherapy undergoing active investigation is combining CTLA-4 blockade with cytokine therapy. IL-2 therapy has been used as adjuvant treatment for melanoma and renal cell carcinoma with benefit in a small subset of patients [225]. IL-2 stimulates T-cell activation, as does CTLA-4 blockade, but through different mechanisms. A phase I/II dose escalation/expansion trial combining ipilimumab with IL-2 was conducted in metastatic melanoma patients. The trial demonstrated a 22 % (5/36) tumor response rate and toxicity similar to prior ipilimumab studies [98]. A phase II trial examining intratumoral injection of IL-2 combined with ipilimumab is currently underway (NCT01480323). There are multiple ongoing trials examining the combination of ipilimumab and high-dose interferon alpha, the cytokine therapy used most frequently as adjuvant therapy in melanoma (NCT01274338 NCT01708941, NCT00610857). GM-CSF has been used in combination with ipilimumab in a phase I dose escalation trial in CRPC demonstrating an immunologic response to treatment as well as a favorable PSA response in the highest dosing cohort (ipilimumab 3 mg/kg and GM-CSF 250 mg every 4 weeks) with expected toxicities [226]. Additional trials of ipilimumab and GM-CSF in CRPC and melanoma are currently underway (NCT01134614, NCT01530984).
6.4.3 Combination Checkpoint Blockade
There is ample preclinical data supporting dual checkpoint blockade in murine cancer models [58, 220, 227–230]. Based on these principles, investigators have initiated trials of dual checkpoint blockade in humans.
Preliminary phase I results of combination of nivolumab (PD-1 blocking mAb) and ipilimumab (CTLA-4 blocking mAb) in patients with advanced melanoma demonstrate the potential of this combination [209]. The trial treated 86 patients with concurrent (n = 53) dose escalation of the two agents or sequenced treatment (n = 33) with nivolumab in patients previously treated with ipilimumab. In the concurrent arm, treatment was dosed at 0.3 mg/kg of nivolumab and 3 mg/kg of ipilimumab (cohort 1), 1 mg/kg of nivolumab and 3 mg/kg of ipilimumab (cohort 2), 3 mg/kg of nivolumab and 1 mg/kg of ipilimumab (cohort 2a), 3 mg/kg of nivolumab and 3 mg/kg of ipilimumab (cohort 3). Dose-limited toxicity was observed in cohort 3; therefore, cohort 2 was treated as the maximum tolerated dose. The concurrent treatment, perhaps not surprisingly, demonstrated considerably higher rates of adverse events than previous trials of either drug in monotherapy. Treatment-related adverse events were noted in 93 % of patients, serious treatment-related adverse events (≥grade 3) were seen in 53 % of patients, and 21 % of patients discontinued therapy secondary to these toxicities. The types of irAEs observed were similar to those seen in both nivolumab and ipilimumab monotherapy trials. That being said, the adverse events were reportedly well managed with immunosuppressant medication and hormonal replacement therapy (for endocrinopathies) and there were no treatment-related deaths observed. In the concurrent arm, 21 of the 53 patients (40 %) were noted to have a response by WHO criteria (the primary endpoint) with the suggestion of a higher response rate when irRC and unconfirmed responses are included. Remarkably, 16 (76 %) of those with an objective response had a tumor reduction of 80 % or more with five complete responses noted.
In the sequenced therapy arm, patients previously treated with ipilimumab were given nivolumab at 1 or 3 mg/kg. The majority of patients (73 %) had progressed on prior ipilimumab therapy. Treatment with nivolumab in sequence was better tolerated than the concurrent therapy with 18 % of patients exhibiting serious treatment-related adverse events (≥grade 3). Objective responses were seen in six of 30 (20 %) evaluable patients with four of the six responses comprising greater than 80 % reduction in tumor volume. Interestingly, no definitive correlation between tumor PD-L1 expression and treatment response could be made in either arm of the trial [209]. A phase III trial comparing the combination of each therapy individually has been designed (NCT01844505).
6.5 Other Checkpoint Pathways Under Development
6.5.1 Lymphocyte Activation Gene-3 (LAG-3)
Lymphocyte activation gene-3 (LAG-3, CD223) is an additional immune coinhibitory checkpoint molecule under investigation for therapeutic purposes in cancer. LAG-3 was first discovered in 1990 on activated T-lymphocytes and NK cells [231]. LAG-3 is structurally similar to CD4 and, like CD4, binds to MHC II complexes on antigen presenting cells (APCs), but with greater affinity [232]. While some early functional data from experiments is mixed, it appears that LAG-3 plays a predominantly inhibitory role in T-cell activation, while promoting APC activation at the same time [198, 233–236].
LAG-3 is expressed on a subset of Treg cells that secretes immunosuppressive cytokines and is more potent that other LAG-3− negative cells of the Treg phenotype (CD4+, CD25highFoxP3+). They are preferentially expanded in patients with cancer [237]. LAG-3 ligation on CD8+ lymphocytes inhibits lymphocyte function and proliferation, independent of Tregs [18]. Notably, high expression levels of LAG-3 are seen on tumor infiltrating lymphocytes and, like PD-1, appear to represent an anergic phenotype [238, 239]. In contrast to its coinhibitory function on T-cells, when soluble LAG-3 binds MHC II complexes on dendritic cells, it promotes activation and maturation [236].
Just as with CTLA-4 and PD-1 pathways, tumor cells are able to utilize the LAG-3 pathway to escape host immunity. MHC class II molecule (LAG-3 ligand) expression is sometimes upregulated to varying degrees in a variety of cancers and can be associated with a worse prognosis [198, 240, 241]. Increased expression of LAG-3 on TILs, corresponding with increased CD8+ T-cell anergy, has been noted in Hodgkin lymphoma, melanoma, and ovarian cancer [242, 243]. Additionally, MHC class II expressing melanoma cells (but not MHC class II negative cells) were resistant to FAS-mediated apoptosis when exposed to LAG-3 transfected cells or soluble LAG-3, indicating a bidirectional signaling in the LAG-3 pathway that effects both lymphocytes and tumor cells [244].
Removing or blocking the LAG-3 pathway improves immune-mediated antitumor effects. Blocking LAG-3 with mAbs has been shown to increase CTL expansion and improved CD4+ lymphocyte cytokine production [245]. In melanoma, anti-LAG-3 mAb blockade improved the antitumor function of tolerized CD8+ lymphocytes when coupled with a viral cancer vaccine [246]. In murine cancer models, PD-1−/− LAG-3−/− knockout mice were capable of rejecting tumors that PD-1 or LAG-3 alone knockout mice could not [227]. It is worth noting that LAG-3−/− knockout mice display a very mild phenotype, similar to PD-1−/− knockout mice, while PD-1−/− LAG-3−/− knockout mice develop lethal autoimmunity at about 10 weeks of age, underscoring the potential toxicity of dual blockade therapy [227, 229, 247]. Similar to the knockout mice, dual mAb blockade of PD-1 and LAG-3 was able to cause complete regression in several established tumor models in mice, while blockade of the individual receptors was not [227].
Since LAG-3 binding of MHC II complexes on APC promotes activation and maturation of the APC, soluble LAG-3 protein has been tested as an immunoadjuvant in cancer. Theoretically, the unbound LAG-3 can promote APC activity while, at the same time, prevent LAG-3-mediated T-cell inhibition through competitive binding. Supporting this, soluble LAG-3 in the serum of breast cancer patients was associated with improved survival [248]. Based on these findings, a fusion protein of the extracellular portion of LAG-3 and the Fc portion of IgG1 was recognized as IMP321 [249]. IMP321 has been tested as a vaccine immunoadjuvant where it was well tolerated and produced encouraging immunologic results [250]. IMP321 has also undergone testing as monotherapy in a phase I dose escalation trial in 21 patients with advanced renal cell carcinoma. The drug produced no significant adverse events and was associated with significantly more disease stability at higher dosing [251]. More recently, IMP321 was tested at two different doses in a phase I trial together with gemcitabine in 12 patients with advanced pancreatic cancer. IMP321 again did not produce significant adverse events but also failed to show any change in immunologic markers after therapy was given [252]. A phase I/II trial of IMP321 along with peptide vaccines in melanoma patients is underway (NCT01308294).
6.5.2 4-1BB
4-1BB (CD137), unlike the inhibitory molecules CTLA-4, PD-1, and LAG-3, is a costimulatory molecule. It is a member of the tumor necrosis factor receptor (TNFR) superfamily that is inducibly expressed on activated CD8+ and CD4+ lymphocytes (including Tregs), NK cells, dendritic cells, macrophages, neutrophils, and eosinophils, as well as in some tumor tissue [253, 254]. The 4-1BB receptor is bound by the 4-1BB ligand (4-1BBL) expressed on antigen presenting cells [254, 255]. 4-1BB functions as a costimulatory signal after a T-cell receptor is bound by an antigen-MHC ligand along with CD28 costimulation to promote CD4+ and CD8+ lymphocyte proliferation, activation, and protection against activation-induced cell death [256–259]. 4-1BB ligation is able to costimulate CD8+ lymphocytes to activation even in the absence of CD28-B7-1/B7-1 signaling and prevent or reverse established anergy in lymphocytes [260, 261]. Additionally, 4-1BB appears to function across both the innate and adaptive immune system as it is able to increase the activity of NK cells which, once activated, are further able to stimulate lymphocyte function [254, 262]. 4-1BB also appears to be functionally important in inhibiting Treg function and promoting antigen priming by dendritic cells [253]. Interestingly, 4-1BB activation via agonistic mAbs is able to prevent or treat antibody-mediated autoimmunity in mouse and primate models by increasing CD4+ (but not CD8+) lymphocyte anergy, a process that is not completely understood [263–265].
Preclinical data with agonistic 4-1BB mAbs has demonstrated a robust antitumor effect. In multiple mouse models, mAb treatment has led to increased tumor-specific CD8+ lymphocyte response and substantial tumor regression [256, 258, 266, 267]. Additionally, melanoma cells transfected to express 4-1BB agonist single-chain Fv fragments and given to mice as an autologous tumor cell vaccine led to rejection of poorly immunogenic tumors [268]. Treatments were well tolerated in animal models, although polyclonal T-lymphocyte accumulation in the liver was noted [269]. Combination of agonist 4-1BB mAb treatment with immunotherapy appears to function synergistically with immunotherapy and chemotherapy [230, 270–273]. To further test its efficacy and safety, one 4-1BB mAb, BMS 663513, was tested in primates along with a prostate-specific antigen DNA vaccine where it demonstrated encouraging immunologic results [253].
Two mAbs have moved into clinical testing in humans. Urelumab (BMS-663513; Bristol Myers Squibb, New York, NY) is a fully human agonist 4-1BB mAb [274] that was given to advanced cancer patients in a dose escalation trial. Initial results from 83 patients with melanoma (54 patients), renal cell carcinoma (15 patients), ovarian cancer (13 patients), and prostate cancer (1 patient) who were given 0.3–15 mg/kg of the mAb with expansion cohorts at the 1, 3, or 10 mg/kg level of dosing have been reported. Results revealed that there were significant toxicities including grade 3 or 4 transaminitis in 11 % and grade 3 or 4 neutropenia in 5 % of patients. There were three objective partial responses in melanoma patients and several other patients with stable disease along with increased levels of peripheral activated T-lymphocytes and interferon in posttreatment biopsies [274]. A phase II trial in advanced melanoma was conducted; however as the incidence of grade IV hepatitis was higher than expected, the trial was terminated. Several other trials were terminated at that time. Two phase I trials are currently enrolling patients in which urelumab is given as monotherapy in advanced solid malignancies or non-Hodgkin lymphoma (NCT01775631) and in combination with rituximab in non-Hodgkin lymphoma or chronic lymphocytic leukemia (NCT01775631). A second drug, PF-05082566 (Pfizer, New York, NY), is currently in phase I trials as monotherapy in solid tumors or in combination with rituximab in non-Hodgkin lymphoma (NCT01307267).
Related posts:
 Gene Therapy and Virus-Based Cancer Vaccines
Gene Therapy and Virus-Based Cancer Vaccines
 Psychoneuroendocrinoimmunotherapy of Cancer
Emerging Biomarkers During Clinical Development of Anti-CTLA4 Antibody Therapy
Psychoneuroendocrinoimmunotherapy of Cancer
Emerging Biomarkers During Clinical Development of Anti-CTLA4 Antibody Therapy
 Polarization of Tumor Milieu: Therapeutic Implications
Polarization of Tumor Milieu: Therapeutic Implications
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


