Fig. 8.1
Distribution of tumor histopathology shown as a percentage of total ovarian tumors in SEER database study (From Brookfield et al. [4]; with permission)
Table 8.1
Histologic classification of ovarian tumors
Germ cell tumors |
Undifferentiated |
Dysgerminoma |
Embryonic |
Teratoma |
Embryonal carcinoma |
Extraembryonic |
Endodermal sinus tumor |
Choriocarcinoma |
Sex cord–stromal tumors |
Granulosa-theca cell |
Sertoli-Leydig cell tumor |
Epithelial tumors |
Serous cystadenoma/cystadenocarcinoma |
Mucinous cystadenoma/cystadenocarcinoma |
Miscellaneous |
Lymphoma |
Leukemia |
Polyembryoma |
Ovarian myxoma |
Mesothelioma |
Gonadoblastoma |
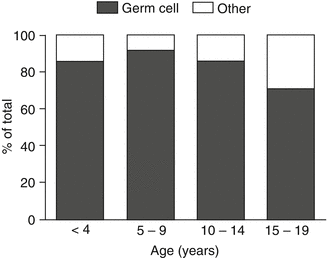
Fig. 8.2
Germ cell tumor shown as a percent of total ovarian tumors by age group (From Brookfield et al. [4]; with permission)
Malignant Germ Cell Tumors
Germ cell tumors (GCTs) arise from the fetal yolk sac: Their heterogeneity stems from the variations in the developmental age, histopathology, and malignant potential of the primordial germ cell. GCTs account for typically 75–80 % of all neoplastic pediatric ovarian masses in most series [3, 4]. All GCTs arise from the primordial germ cell but can express different tumor markers and have different malignant potentials, resulting from the differences in the stage of germ cell development at tumorigenesis and the tumor microenvironment secondary to the location of the clone. The most undifferentiated GCTs are dysgerminomas. Germ cells that undergo some degree of differentiation before becoming neoplastic either differentiate into embryonic tumors (embryonal carcinoma or teratoma) or extraembryonic tumors (endodermal sinus tumors or choriocarcinoma) as is shown in Figure 8.3 [26].
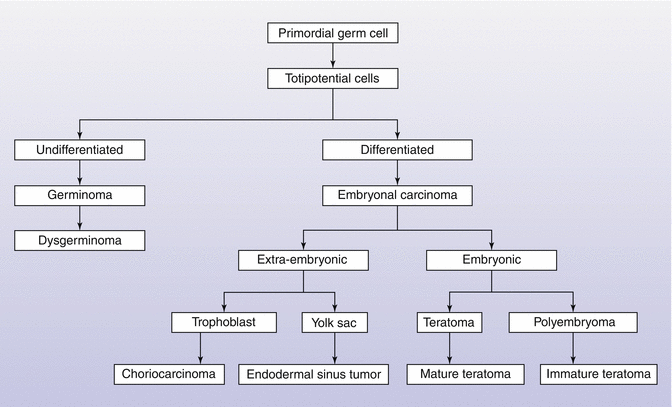
Fig. 8.3
Histologic derivation of ovarian germ cell tumors
Teratomas
Teratomas are the most common ovarian tumors and are either mature (benign), immature (contain malignant potential), or malignant. The mature teratoma is composed of mature representative tissue from all three germ layers: ectoderm, mesoderm, and endoderm. Immature teratomas have a gross similar appearance to that of a mature teratoma but uniquely have various foci of immature tissues present, usually neuroepithelium. Immature teratomas in children behave in a malignant fashion only if they are clinically advanced (advanced stage) or contain foci of malignant germ cell elements (yolk sac tumor). Clusters of yolk sac tumor may be very small and associated intimately with immature neural tissue and thus do not stain avidly for AFP and are overlooked. These foci of immature elements are assumed to be responsible for the metastatic potential of an immature teratoma.
All grading systems quantify the degree of immaturity in the lesion. Immature tumors are graded from I to III based on the amount of neuroepithelium or the degree of immature elements present in the mass (see Table 8.2). Grade I is the most differentiated with the least risk of malignancy, and grade III is the least differentiated with the greatest risk of malignancy. The histologic level of differentiation is determined by the presence of immature elements: the more immature neural elements result in a higher histologic grade which correlates with a more aggressive biology of the tumor in adults [27]. This grading of the extent of immature lesions has shown potential prognostic significance only in adults, and chemotherapy is required to reduce recurrences in adults with this aggressive phenotype. In contrast, an intergroup study of Children’s Oncology Group and the Pediatric Oncology Group examined the need for adjuvant therapy in children and concluded that surgery alone is an adequate treatment for an immature teratoma, even in the presence of an elevated AFP suggestive of a component of YOLK Sac tumor [28]. Table 8.3 correlates serum tumor markers with underlying histopathology. Five-year survival for pure immature teratomas by histologic grade was 81, 60, and 30 % for grade I to III, respectively [29]. Survival among pure immature teratomas has shown 74 % 5-year survival for all grades of stage I and 38 % survival for stages II and III [27]. Many teratomas are associated with the presence of peritoneal implants of mature glial tissue, gliomatosis peritonei, which is a distinct entity and does not change tumor stage or alter prognosis.
Table 8.2
Grading of immature ovarian teratomas
Grade 0 | All tissues mature; no mitotic activity |
Grade 1 | Some immaturity but with neuroepithelium absent or limited to one low-magnification field; no more than one focus in a slide |
Grade 2 | Immaturity present; neuroepithelium common but no more than three low-power microscopic fields in one slide |
Grade 3 | Immaturity and neuroectoderm prominent; no more than four low-power fields per section |
Table 8.3
Ovarian tumor markers
Pathology | Tumor markers | Incidence of bilaterality |
|---|---|---|
Teratoma | ±AFP | 10 % |
Dysgerminoma | LDH-1 | 15 % |
Endodermal Sinus Tumor | AFP | Low |
Choriocarcinoma | β-HCG | Low |
Epithelial tumors | CA-125 | 15 % |
Germinomas
Malignant dysgerminomas arise from the primordial undifferentiated germ cells and are the most common pure germ cell malignancy in the ovary, accounting for 50 % of malignant ovarian GCTs [30]. They can occur in dysgenetic gonads, those abnormal gonads containing gonadoblastoma [31], or phenotypic females with karyotypic abnormalities. The majority of pure dysgerminomas present as stage I disease and are highly responsive to chemotherapy and radiation and are histologically identical to a seminoma in males. Bilateral disease occurs in 10–15 % of cases [32]. The 10-year survival rates for a dysgerminoma confined to one ovary and treated with surgery is 92 %; however, there is a noted 17 % recurrence rate and 6 % mortality rate [32].
Yolk Sac Tumor (Endodermal Sinus Tumor)
Endodermal sinus tumor, also called a yolk sac tumor, is of extraembryonic germ cell origin and is the second most common pure GCT [33]. It is typically associated with elevations of AFP [33] (see Table 8.3) and has an aggressive biology with peritoneal spread and metastases to the liver, lung, and brain [34, 35]. There are four general histopathologic patterns but these patterns are not clinically relevant. The “pseudopapillary or festoon” pattern and “microcystic or reticular” pattern are the most common. The “solid” pattern is usually found only focally and can resemble embryonal carcinoma; the “hepatoid” pattern closely resembles fetal liver and is a variant of the “solid” pattern. The fourth pattern is the “polyvesicular vitelline” pattern.
Endodermal sinus tumors can be associated with gonadoblastomas in some cases. Gonadoblastomas are benign tumors typically and occur exclusively in phenotypic females with at least a portion of the Y chromosome; thus they present clinically as male pseudohermaphrodites and have dysgenetic gonads. Germinomas frequently develop with gonadoblastomas [36].
Mixed Cell Tumors: Embryonal Carcinoma and Choriocarcinoma
Mixed GCTs contain multiple malignant cell types and carries the phenotypic biology and diagnosis of the “worst” acting cellular element. They constitute up to 30 % of malignant GCTs [37]. Both embryonal carcinoma and choriocarcinoma are rare in isolation but are more commonly seen in mixed tumors: both produce β-HCG, and embryonal carcinoma can produce precocious puberty. The two histopathologic forms of embryonal carcinoma—the pseudotubular and papillary patterns—can be confused with yolk sac tumors, but the cells are AFP negative and lack the eosinophilic hyaline globules characteristic of yolk sac tumors. Embryonal carcinoma stains positive for CD30 by immunohistochemical staining.
Choriocarcinoma also rarely occurs as a pure lesion, and when it appears in infants, it represents metastases from maternal or placental gestational trophoblastic primary tumor [38, 39]. These tumors are very friable and hemorrhagic and require the presence of two cell types—cytotrophoblasts and the syncytiotrophoblasts—which stain for β-HCG for diagnostic confirmation.
Polyembryoma is a very rare malignant lesion with a poor prognosis, occurring as a component of a mixed lesion. Both AFP and β-HCG can be elevated and also this tumor presents often with symptoms of precocious puberty [40].
Non-Germ Cell Tumors
Non-germ cell tumors include both sex cord–stromal tumors (stromal histology) and the epithelial tumors (epithelial histology). Tumors of epithelial or stromal cells of origin each accounts for the remaining 15–20 % of ovarian malignancies that are not germ cells.
Sex Cord–Stromal Tumors
Sex cord–stromal tumors include Sertoli–Leydig cell tumors and juvenile granulosa cell tumors, arising from the stromal components of the ovary and are hormonally active. Juvenile granulosa cell tumors, constituting 10–20 % of pediatric ovarian tumors, have a malignant potential although they follow a benign clinical course in children unlike adults. The pure granulosa tumor is highly malignant, mixed tumors are less malignant, and the pure thecoma is benign. The majority (>95 %) develop in postpubertal girls with estrogen excess and can be associated with pseudo-precocious puberty in 80 % of girls with breast enlargement, vaginal bleeding, and galactorrhea [41, 42]. Menstrual irregularities and swelling are the presenting symptoms. Most cases are limited to the ovary at presentation with <5 % being bilateral so that a unilateral salpingo-oophorectomy and appropriate staging is usually adequate therapy. Serum inhibin levels are associated with determining recurrence. Prognosis is excellent with 84–92 % survival.
Sertoli–Leydig cell tumors, arrhenoblastomas, are rare and account for <0.5 % of all malignant ovarian neoplasms [43]. The less well-differentiated and retiform histologies are most commonly found in children, most often localized to the ovary, and represent a low-grade malignancy. Clinically they present with androgen excess, precocious puberty in young children, and hirsutism or virilization in adolescents. An elevated AFP is associated with this tumor [44]. Prognosis depends on the stage and degree of differentiation of the tumor; usually it is a low-grade malignancy, so a unilateral salpingo-oophorectomy with staging is sufficient. Multiagent chemotherapy is utilized for advanced, metastatic, or recurrent disease [45].
In stromal cell tumors, mitotic activity in conjunction with tumor stage correlates with patient outcome. Tumors with >20 mitoses per high-power field (HPF) had an event-free survival (EFS) that was half of those with <20 mitoses per HPF [46]. Those tumors with high proliferative activity are the high risk category and difficult to treat.
Epithelial Tumors
In adults, epithelial tumors represent the majority of ovarian tumors, but in contrast less than 20 % of those in childhood are derived from the surface epithelium of the ovary and are rare to present before menarche [47]. The histologic subtypes in children are limited to serous or mucinous tumors which can be characterized as benign, borderline (of low malignant potential), or malignant. Approximately 84 % of epithelial lesions in two case series were benign or of low malignant potential [6, 48].
Borderline epithelial tumors are defined as neoplasms with varying nuclear atypia that lacks stromal invasion of the ovary and occur three times more frequently in children than adults [39]. Cystadenomas are usually benign with 7 % being borderline and 4 % being malignant [49–52]. Borderline tumors are indolent and can be bilateral. They can present locally extensive and proper workup would require applying the proper operative staging of epithelial tumors using adult guidelines since they are an epithelial histology, as opposed to the pediatric guidelines designed for germ cell tumors.
Metastatic Disease to the Ovary
Molecular Pathology of Ovarian Tumors
Mature Teratomas
Ovarian mature GTCs are thought to arise primarily from benign germ cells in meiosis and not from a malignant transformation of the germ cell. The entry into meiosis of primordial germ cells is a gradual process that continues until birth. Over 95 % of mature ovarian teratomas are karyotypically normal with only 5 % showing gains of a single whole chromosome [58, 59]. Analysis of molecular loci has shown that the majority has entered but not completed meiosis and thus is diploid [58].
Immature Teratomas
Immature and mature teratomas appear to have a different mechanism of origin and thus represent different biologic entities rather than occurring along the same spectrum of maturation. The origins of immature teratomas appear to come from meiotic stem cells and others from mitotic cells, implying a failure of early meiotic arrest [60], whereas mature teratomas originate from germ cells entering but not completing meiosis, a normal process of maturation. The frequency of chromosomal abnormalities is higher in immature teratomas than mature teratomas. Chromosomal analysis suggests that immature ovarian teratomas of premeiotic origin show a greater malignant potential and deviations in karyotype suggest a worse prognosis [61, 62]. The majority of patients with cytogenetically abnormal immature teratomas have recurrent disease. In contrast those with karyotypically normal immature teratomas have remained disease-free [60–62].
Malignant Ovarian Germ Cell Tumors
Immature teratomas may develop genetic changes found in malignant teratomas which correlate with the histologic malignant transformation of malignant GCTs. Malignant ovarian teratomas are presumed to originate from the malignant transformation of a germ cell. The genetics of malignant ovarian teratomas are similar to those of testicular tumors with similar ploidy and genetic features. Malignant teratomas often are aneuploid and 75 % may contain the isochromosome 12p, i(12p), common in malignant testicular teratomas [63]. Other genetic features include 42 and 32 % have gains of chromosomes 21 and 1q, respectively, whereas 25 and 42 % have loss of chromosomes 13 and 8, respectively [64–66].
The association between sex chromosomal abnormalities and the development of GCT is well established. Individuals with 46XY and 45X/46XY gonadal dysgenesis have a 10–50 % risk of developing a gonadal germ cell tumor [67].
Dysgerminomas, in particular, are associated with abnormal karyotypes in phenotypic females: 46XY as in gonadal dysgenesis or complete androgen insensitivity, or 45X/46XY as in mixed gonadal dysgenesis.
Clinical Presentation
Although rare in presentation, ovarian tumors must be included in the differential diagnosis of abdominal pain or an abdominal mass in female children. Pain is the most common symptom, although the presence of an abdominal mass was frequent and other symptoms are often nonspecific. Precocious puberty and increasing abdominal girth are also suspicious for an underlying ovarian malignancy. The symptoms of abdominal pain and increased girth, with nausea and vomiting, also typify appendicitis, which is the primary diagnosis that causes the majority of these operations being performed on an emergency basis without a complete workup or tumor markers. Exquisite tenderness may suggest peritoneal irritation from rupture or torsion of the mass.
The largest case series of ovarian masses analyzed differences in presentation between those that are benign or malignant masses [5]. Perimenarchal patients tend to have a greater number of benign lesions such as cysts and neoplasms, while those younger patients that are hormonally quiescent are less likely to present with an ovarian mass at all. The greatest percentage of malignancies in all ovarian lesions was in those patients aged 1–8 years, with malignancy being present in up to a quarter of the time. As a result, a higher index of suspicion for malignancy is needed in the patient aged 1–8 years, as indicated by a threefold increase in the odds ratio. Conversely, it is exceedingly rare for a child less than 1 year to have a malignant neoplasm [5, 68, 69].
The presenting symptoms of patients with malignancies can significantly overlap with those with benign conditions. Complaints of an abdominal mass or precocious puberty are statistically more often associated with an underlying malignancy [6, 68, 69]. Precocious puberty in the setting of a mass can be associated with sex cord–stromal tumors, including Sertoli–Leydig cell tumors, and GCTs, such as embryonal carcinoma and polyembryoma. Evidence of unusual hormonal activity is more often associated with either estrogen production from a juvenile granulosa cell tumor or androgen production with resultant virilization from a Sertoli–Leydig cell tumor [70, 71]. Not infrequently, a similar level of hormone production can be seen with autonomously functioning benign follicular cysts [22]. In contrast over 50 % of epithelial tumors in children present as asymptomatic masses incidentally detected [6].
A recent largest single institution review analyzed the features of benign ovarian masses compared to malignancies to best unravel the diagnostic dilemma of an ovarian mass on presentation. The findings and odds ratios suggestive of a potential malignancy included masses in those aged 1–8 years of age, a chief complaint of a mass or precocious puberty, imaging findings of a mass greater than 8 cm, or a mass that is solid in character (shown in Table 8.4) [5]. Laboratory investigations showing elevated β-HCG, AFP, or cancer antigen 125 (CA-125) were worrisome for malignant neoplasm but are not definitive if normal.
Table 8.4
Criteria for suspicious malignant ovarian masses
Adnexal masses with extra-ovarian spread |
Ultrasonically suspicious masses >8 cm |
Thick cyst wall by ultrasound |
Lengthened utero-ovarian ligament |
Numerous vessels starting from the mesovarium with a comb-like pattern |
Peritoneal metastasis |
External ovarian vegetations |
Intracystic vegetations |
Management Strategy
Evaluation
The work-up involves comprehensive exam, preoperative imaging, and testing for serum tumor markers, and if they are abnormal, then consult with a pediatric oncologist. Preoperative imaging will often reveal a pelvic mass, easily detected by US, computed tomography (CT) scan, or magnetic resonance imaging (MRI), depending on the initial complaints prompting the radiologic evaluation. Malignant masses were generally thought to be greater than 5 cm [48, 68], whereas others advocate the use of >8 cm to be the size cutoff for concern of malignancy [72]. In a case series, all malignancies exceeded 6 cm, whereas ≥8 cm allowed for a statistically significant odds ratio with the greatest accuracy (Table 8.4). The caveat is that using the 8-cm cutoff, two of 46 malignancies (4.3 %) would have been missed by size alone [5].
Radiologic studies are not always able to distinguish benign lesions from malignancies. Classically, solid masses are thought to have a higher risk of malignancy, although heterogeneous and cystic masses can also harbor underlying malignant neoplasm [7]. Solid masses, either by ultrasound or by CT scan had the highest frequency (33–60 %) of malignancy; solid features were only evident in 14–15 % of all malignancies [5]. Conversely, the majority of radiologic reads identified either a heterogeneous or cystic mass, which were associated with malignancy in only 15–20 % and 4–5 % of the time, respectively, which is consistent with previous reports [73]. While findings of a cystic lesion may be relatively reassuring, heterogeneous masses should still be approached with a heightened suspicion for malignancy, especially if additional concerning factors are present.
If an ovarian mass >8 cm is seen on preoperative imaging, serum levels of AFP, β-HCG, and CA-125 should be evaluated before surgery, and a chest CT scan should be obtained to rule out metastatic disease. If signs of precocious puberty are apparent, then FSH, LH, estradiol, and lactate dehydrogenase (LDH) serum levels should be obtained.
Tumor Markers
β-HCG, AFP, and CA-125 are all potential serum tumor markers for ovarian malignancies. GCTs will often produce AFP and/or β-HCG, depending on their subgrouping (see Table 8.3) [74]. Dysgerminomas can result in elevations of serum lactic dehydrogenase (LDH), β-HCG, or CA-125 [74]. Yolk sac and Sertoli–Leydig cell tumors are both associated with AFP elevations. CA-125 is most commonly associated with epithelial type tumors [6]. However, the caveat is that tumor markers do not exclude the possibility of malignancy when they are not elevated.
Thus, tumor markers are not definitive measurements preoperatively but are of greatest value, if elevated, in the ability to monitor postoperatively for complete resection of disease, as well as to detect relapse [75, 76]. Tumor markers should be drawn preoperatively whenever possible, but elevations can still be detected immediately postop due to the relatively long half-lives of both AFP and β-HCG [75].
Related posts:
 Psychological Aspects of Hereditary and Non-hereditary Ovarian Cancer
Psychological Aspects of Hereditary and Non-hereditary Ovarian Cancer
 Management of Patients with Early-Stage Ovarian Cancer
Management of Patients with Early-Stage Ovarian Cancer
 Ovarian Cancer Screening and Early Detection
Ovarian Cancer Screening and Early Detection
 Treatment of Advanced Stage Ovarian Cancer
Treatment of Advanced Stage Ovarian Cancer
 Pathology of Non-epithelial Malignancies of the Ovary
Pathology of Non-epithelial Malignancies of the Ovary
 Surface Epithelial Tumours of the Ovary
Surface Epithelial Tumours of the Ovary
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


