K is expressed on red cells by about the tenth week of life (Marsh and Redman 1990). Nine per cent of the English population are K (K1) positive, having the genotype KK or Kk; the frequency of K is 0.046 and that of k (K2), 0.954. (Throughout this chapter, frequencies of alleles in white people are taken from Race and Sanger 1975.) In black people in the USA the frequency of K positives is about 1.5% (Stroup et al. 1965) and in Japanese is only about 0.02% (Hamilton and Nakahara 1971).
Other sets of alleles are closely linked to K1 and K2 but only two of these sets, Kpa, Kpb and Kpc (K3, K4, K21) and Jsa and Jsb (K6, K7) are known to be clinically important. In white people, Kpb and Jsb have a very high frequency. Although Jsa is rare in white people, it is not uncommon in black people. In the extremely rare null phenotype K0, no Kell system antigens are present on red cells. Ku (K5) is found on all red cells except those that are K0.
In the numerical notation for Kell, as for Rh and other systems, phenotypes are designated according to reactions with particular antibodies; for example, a sample reacting negatively with anti-K is designated K: −1; if the sample has also reacted positively with anti-k, it is designated K: −1, 2.
Numbers of K and k Antigen Sites
Using both polyclonal and monoclonal radio-iodinated anti-K, the number of K sites on KK red cells was found to be 4000–6100 and on Kk cells 2500–3500 (Hughes-Jones and Gardner 1971; Jaber et al. 1989). Using four monoclonal antibodies reacting with epitopes absent from K0 red cells, 2000–4000 epitopes were detected per red cell; with Fab fragments of the same monoclonals, estimates were higher, namely 4000–8000 sites in three out of four cases and 18 000 in the fourth (Parsons et al. 1993). Using ferritin-labelled anti-IgG and polyclonal IgG anti-k, the number of k sites, on both Kk and kk cells, was found to be 2000–5000 (Masouredis et al. 1980a).
Molecular Biology and Chemistry of Kell Antigens
The glycoprotein that carries Kell system antigens (CD238) is encoded by a gene on chromosome 7 at 7q33. It has a molecular weight of 93 kDa and comprises 732 amino acids. It is a type II membrane protein with its amino-terminal domain in the cytosol, a single membrane-spanning domain and a large extracellular carboxy-terminal domain of 665 amino acids. The extracellular domain contains 15 cysteine residues, suggesting that this part of the molecule is highly folded, consistent with its sensitivity to reducing agents. One of these residues (Cys72) forms a disulphide bond with the Xk protein. There are five potential glycosylation sites in the extracellular domain. The molecule is a member of the M13 group of zinc endopeptidases and its preferred substrate is big endothelin-3. Proteolytic cleavage of big endothelins releases bioactive peptides with diverse physiological functions including vasodilation effects. The molecular bases of most Kell system antigens are known and all result from single amino acid substitutions (Table 6.1; Redman et al. 1999; Daniels 2002; Lee et al. 2006; Daniels et al. 2009). The Ko and Kmod phenotypes result from a diverse array of inactivating mutations in the KEL gene (Lee et al. 2001; Lee et al. 2003a; Wester et al. 2005). A model of the Kell protein based on the crystal structure of neutral endopeptidase 24.11 (NEP) indicates that the extracellular domain of the protein has two globular domains rich in alpha helix. The domain closest to the lipid bilayer encompasses the enzyme active site, whereas the domain furthest from the membrane contains all the amino acids whose substitution is related to the expression of blood group antigens (Lee et al. 2003b; Colour Plate 6.1).
K/k polymorphism is determined by methionine at position 193 in K rather than threonine, as in k (Colour Plate 6.1). The ability to distinguish K from k in a polymerase chain reaction (PCR) method that can be applied to DNA samples obtained from amniotic fetal cells is proving useful in predicting haemolytic disease in pregnant women with anti-K (Lee et al. 1995; Van der Schoot et al. 2003; see also Chapter 12).
Using the monoclonal antibody-specific immobilization of erythrocyte antigens (MAIEA) technique (see Chapter 8), at least five spatially distinct regions could be distinguished on the Kell glycoprotein; for instance, one containing K, k, Ula, K22 and, possibly, Jsa and Jsb, and another containing Kpa, Kb and Kpc (Petty et al. 1994). Kell antigens are destroyed by treating red cells with a reagent ‘ZAPP’, containing 0.1 mol/l of dithiothreitol (DTT) and 0.1% papain (Branch and Petz 1980), or by treatment with 2-aminoethylisothiouronium bromide (AET). K0 red cells, occurring naturally or produced by treating red cells of common groups with AET, may be useful in demonstrating that antibodies are or are not recognizing antigens within the Kell blood group system and may also be useful in recognizing antibodies outside the Kell system when these are present together with Kell antibodies (Advani et al. 1982). However, some antigens outside the Kell system are also inactivated by AET (see Chapter 8).
Kell antigens are inactivated after the combined treatment of red cells with trypsin and chymotrypsin; treatment with either of these enzymes separately enhances the expression of Kell antigens (Judson and Anstee 1977).
Red cells can acquire K-like antigens; the red cells of a K-negative patient with Streptococcus faecium septicaemia were shown to react with anti-K. K-negative red cells became agglutinable by anti-K when treated in vitro with the streptococcus isolated from the patient. Jk(b−) cells also became agglutinable by anti-Jkb when this organism was disrupted and used to treat the cells (McGinniss et al. 1984).
Kell Antigens on Other Cells
Using immunofluorescence flow cytometry, K and k antigens were not detected on lymphocytes, monocytes or granulocytes (Dunstan 1986) or on platelets (Dunstan et al. 1984). However, Kell protein could be detected in testis, skeletal muscle and lymphoid tissues and was coisolated with Kx protein in skeletal muscle (Russo et al. 2000).
The Antigen Kx
Kx is expressed most strongly on red cells that lack Kell antigens, i.e. K0 red cells. Similarly, red cells treated with AET (see above) have a high expression of Kx (Advani et al. 1982).
Kx is the only antigen in the Kx system; its inheritance is determined by an X-borne recessive gene Xk at Xp21. The gene encodes a protein of 444 amino acids of a molecular weight of 51 kDa with 10 putative transmembrane domains, a very short (four amino acids) cytosolic amino-terminal domain and a larger (71 amino acids) cytosolic carboxy-terminal domain (Ho et al. 1994). A single extracellular cysteine residue (Cys 347) on the fifth extracellular loop forms a disulphide bond with Cys72 of the Kell protein. The function of Kx protein is unknown but its structure and covalent linkage to a type II membrane protein (Kell protein) is very reminiscent of the family of heterodimeric amino acid transporters (Southcott 1999; Palacin and Kanai 2004).
Kx is present on all red cells except those belonging to the very rare McLeod phenotype, on which all Kell antigens are depressed. Subjects with this phenotype have various other characteristics, which together constitute the McLeod syndrome: acanthocytic red cells, elevated serum creatine kinase, muscle weakness, cardiomyopathy and neurological defects resembling those of Huntington’s disease (Marsh et al. 1981; for a recent review, see Jung et al. 2007). Consistent with this the Xk gene is expressed in skeletal muscle and brain (Ho et al. 1994).
The McLeod phenotype can result from deletion of that part of the X chromosome that carries Xk but other inactivating mutations have also been described (Russo et al. 2002). A minority of patients with the X-linked type of chronic granulomatous disease also have the McLeod phenotype and in these the region of X that is deleted includes the locus for X-linked CGD as well as the Xk locus and these patients may make anti-Kx upon transfusion. Rarely, female heterozygotes have the McLeod phenotype, most likely as the result of random X-chromosome inactivation (reviewed in Jung et al. 2007).
Antibodies of the Kell and Kx Systems
Anti-K
Naturally Occurring Anti-K Is Rare
Although an example that was IgG has been described (fourth edition, p. 344), most examples of naturally occurring anti-K are IgM, usually reacting best at room temperature and, in some cases, first found after an illness and disappearing following the patient’s recovery (Tegoli et al. 1967; Kanel et al. 1978; Judd et al. 1981). In one case, the antigenic stimulus was an infection with an uncommon strain of Escherichia coli O125: B15. A cell-free filtrate of a culture of the organisms inhibited the anti-K (and the anti-A) in the serum of the patient, who was a newborn infant. Both the anti-K and the anti-A had disappeared by the time the infant was 3 months old (Marsh et al. 1978). One patient had pulmonary lesions suggestive of tuberculosis and in another the relevant organisms were unidentified (Tegoli et al. 1967; Kanel et al. 1978). Some strains of Campylobacter jejuni and Campylobacter coli, major aetiological agents of gastrointestinal infections in humans, carry surface sites reactive with anti-K (Wong et al. 1985).
Immune anti-K is the commonest immune red cell antibody outside the ABO and Rh systems, accounting for almost two-thirds of non-Rh immune red cell alloantibodies (see Table 3.6). An estimate of the frequency with which it is formed in K-negative subjects transfused with at least 1 unit of K-positive blood was made by Kornstad and Heistö (1957). In a series of 130 such subjects, 53 were tested at 4–12 months and five had anti-K; of the remaining 77 subjects, tested at 13–37 months, only one had anti-K. The authors concluded that the probability that a K-negative subject transfused with a unit of K-positive blood would develop anti-K was about 1 in 10. If this estimate were correct, K would be eight times less immunogenic than D, but it may be even less immunogenic than this.
Other evidence of the immunogenicity of K comes from two series of experiments: in the first, three spaced injections of K-positive red cells were given to 16 subjects, at least 10 of whom were K negative; none formed anti-K (Wiener et al. 1955). In the second, a series of injections of K-positive, D-positive red cells was given to 31 K-negative, D-negative subjects (FM Roberts, personal communication). Of the 19 subjects who failed to form anti-D, none formed anti-K. Of the 12 who formed anti-D, six formed anti-K but it is well known that subjects who make anti-D have a greatly increased chance of making antibodies to other antigens carried by the immunizing D-positive red cells (see Chapter 3).
Opportunities for alloimmunization to K as a result of pregnancy are relatively common but, assuming that K is 10 times less immunogenic than D, it can be shown that the incidence of HDN due to anti-K in second pregnancies would be expected to be only about 1 in 3500 (seventh edition, p. 405). Data on the frequency with which anti-K is found in pregnancy are given in Chapter 12.
In screening samples for anti-K, Kk red cells are almost always used; as some samples of anti-K can be detected only with KK red cells, the estimates given above for the frequency of anti-K must be too low.
The woman in whose serum anti-K was first discovered (Coombs et al. 1946) had been transfused previously with blood from her husband (information not in the original paper), as had the woman investigated by Chown (1949) who emphasized the moral ‘never transfuse a woman with her husband’s blood’.
Several monoclonal antibodies with K specificity have been produced.
Anti-k
Anti-k was first found in the serum of a recently delivered woman whose infant was mildly affected with haemolytic disease of the newborn (Levine et al. 1949). The rarity of the antibody is accounted for by the rarity of k-negative (i.e. KK) subjects: 0.04–2% in white people in various parts of Europe, 0.005% in black people in the USA and very much rarer in Japanese (see Daniels 1995). A murine monoclonal anti-k has been described (Sonneborn et al. 1983).
Other Antibodies of the K System and Anti-Kx
Anti-Kpa, anti-Kpb, anti-Jsa and anti-Jsb are all rare. The original example of anti-Kpa appeared to be naturally occurring (Allen and Lewis 1957); this antibody and anti-Kpb may also occur as autoantibodies (see Chapter 7); the other antibodies mentioned in this section have been described only as immune alloantibodies. Anti-Ku reacts with all samples except K0 and is found only in K0 subjects (Corcoran et al. 1961).
Subjects with the McLeod phenotype may make two alloantibodies following transfusion, anti-Kx and anti-Km. Km is an antigen present on red cells of common Kell phenotypes but not on K0 red cells or on cells of McLeod phenotype.
Four monoclonal antibodies recognize high-frequency epitopes absent from K0 cells; all four identify the Kell glycoprotein (molecular weight 95.6 kDa); the four epitopes recognized fall into two non-overlapping groups: the first includes Kpbc and K14 and the second, k (Parsons et al. 1993).
An antibody found in patients with the McLeod syndrome, previously known as anti-KL, is a mixture of anti-Kx and anti-Km and is found only after transfusion. Anti-Km reacts with all samples except those that are K0 or Kx negative; anti-Kx, on the other hand, reacts strongly with K0 red cells, which, as already mentioned, possess greatly increased amounts of Kx. Anti-Kx can cause rapid destruction of Kx-positive red cells, associated with haemoglobinuria, but does not interfere with the successful transfusion of granulocytes (Taswell et al. 1976), implying an absence of Kx from such cells.
Serological Characteristics of Immune Anti-K and Anti-k
Anti-K and anti-k are usually IgG. Kell antibodies were found to be solely IgG1 in seven out of eight and 12 out of 14 cases respectively (Engelfriet 1978; Hardman and Beck 1981). As with anti-D, sera containing only IgG anti-K may, when undiluted, agglutinate saline-suspended cells. Many examples of IgG anti-K activate complement but only to the C3 stage, and are non-lytic.
Anti-K may be IgM but only the most potent examples agglutinate red cells suspended in saline; weaker examples sensitize red cells to agglutination by anti-IgM and sometimes to anti-complement.
Anti-K tends to react poorly in low-ionic-strength solution (LISS) and may be relatively difficult to detect with the LISS-IAT, or the low-ionic-strength polybrene method, with or without the IAT, and in the AutoAnalyzer (see Chapter 8).
Studies with 125I-Labelled Anti-K
The equilibrium constants of several examples of IgG anti-K ranged from 0.6 to 4.5 × 1010 l/mol, figures that are 100 times greater than those found with IgG anti-D, indicating that, if anti-K binds monovalently, its intrinsic affinity for K must be much higher than that of anti-D for D. The concentration of anti-K corresponding to an indirect antiglobulin titre of 1 is about the same as for anti-D (Hughes-Jones and Gardner 1971).
Clinical Aspects
Anti-K and much less frequently anti-k can cause severe haemolytic transfusion reactions; anti-Jsa and -Jsb have been known to cause a delayed haemolytic transfusion reaction (DHTR) (for references, see Daniels 1995). Anti-Kpa, and far more rarely -Kpb and -Jsb, have caused severe HDN and milder disease due to anti-Jsa and -Ku has been reported (for references, see Chapter 12). Anti-K has several times been the cause of haemolytic transfusion reactions due to interdonor incompatibility (see Chapter 11 and West et al. 1986). Antibodies of the Kell system, particularly anti-Kpb, may be found as autoantibodies in autoimmune haemolytic anaemia (AIHA); there may be concomitant depression of the patient’s own Kell antigens (see Chapter 7).
Transient depression of Kell antigens, with the development of alloantibodies reacting with a high-incidence Kell antigen or with all but K0 cells has been observed in association with autoimmune thrombocytopenic purpura (Vengelen-Tyler et al. 1987; Williamson et al. 1994). In the second of the two cases, in a subsequent relapse of autoimmune thrombocytopenic purpura (AITP) the red cells became Lu(a− b−) and an antibody against a high-incidence Lu antigen appeared in the serum. In both relapses, antigen changes persisted for many months; LWa and CD44 were also markedly weakened.
The Duffy (Fy) System
The red cells of about 66% of the English population are Fy(a+), belonging to the genotype FyaFya or FyaFyb, the remainder being FybFyb (Cutbush and Mollison 1950).
In white people, the alleles Fya and Fyb have frequencies of 0.425 and 0.557 respectively; a further allele, Fyx, which makes a weak form of Fyb, has a frequency of 0.016. The frequency of the gene Fy, which produces neither Fya nor Fyb, is only 0.002 but in black people in the USA is about 0.7. In parts of tropical Africa the frequency of Fy is 1.0, all the native inhabitants having the phenotype Fy(a− b−) (Mourant et al. 1976). A high frequency of Fy(a− b−) is also reported in the Jarawas of the Andaman islands (Das et al. 2005). Fy(a− b−) is extremely rare in white people and most other races.
The Fy locus is on chromosome 1q21–q25; it was the first blood group gene to be assigned to an autosome in humans (Donahue et al. 1968).
A monoclonal antibody produced in mice by injecting pooled human red cells was found to recognize a unique epitope on the Duffy glycoprotein and was named anti-Fy6 (Nichols et al. 1987).
Fya and Fyb Sites on Red Cells and Nature of the Fya and Fyb Antigens
The number of Fya sites on FyaFya red cells and of Fyb sites on FybFyb red cells was estimated to be 17 000; the number of Fya sites on FyaFyb was estimated to be 6900 (Masouredis et al. 1980b). Using flow cytometry, Woolley et al. (2000) report that Fy antigen expression is significantly higher (by 49 ± 19%) on reticulocytes than on mature red cells.
Using an immunoaffinity technique for isolating antigens in the form of soluble antigen–antibody complexes, the Fya and Fyb antigens were shown to be associated with membrane components of apparent molecular weight of 40 kDa (Moore 1983). Similarly, using immunoblotting of red cell ghosts with a potent anti-Fya, the Fya antigen was located on a glycoprotein of 35–43 kDa, migrating between bands 5 and 6 (Hadley et al. 1984).
Sequencing of cDNA encoding the Duffy glycoprotein (synonym: DARC, Duffy antigen chemokine receptor) revealed a protein of 338 amino acids with an apparent molecular weight of 35 733 (Chaudhuri et al. 1993; Iwamoto et al. 1996). The protein would be predicted to have seven membrane-spanning domains with an amino-terminal extracellular domain of 65 amino acids and a small carboxy-terminal cytoplasmic domain. All of the three potential N-glycosylation sites in the extracellular amino-terminal domain (N16SS,N27SS, N33DS) can be utilized (Czerwinski et al. 2007). The Fya/Fyb polymorphism is due to a point mutation at amino acid 44, there is Asp in Fyb instead of Gly in Fya (Mallinson et al. 1995). The Fy6 epitope has been mapped to a region in the extracellular domain involving residues 22 (Phe), 23 (Glu), 24 (Asp) and 25 (Val), whereas Fy3 is influenced by residues 58 (Asp) and 59 (Asp) in extracellular domains 1, 124 (Arg) in domain 2 and 263 (Asp), 283 (Asp) in domain 4 (Tournamille et al. 2003). A structural model of the Duffy glycoprotein depicting the location of these antigens is provided by Brevern et al. (2005; Colour Plate 6.2). The Fyx (synonym: weak Fyb expression) results from a nucleotide substitution (C265T) resulting in Arg89Cys in the first intracellular loop of the Duffy glycoprotein. The replacement of arginine appears to adversely affect the insertion of the protein into the plasma membrane and the reduced levels of surface protein that result account for the weak expression of Fyb antigen (and Fy3, Fy5 and Fy6) on the red cells (Olsson et al. 1998; Tournamille et al. 1998).
The Fya antigen is destroyed by crude preparations of proteolytic enzymes although not by crystalline trypsin (or neuraminidase); it is destroyed by chymotrypsin (Morton 1962). Fy3, 4 and 5 are not destroyed by papain. Treatment of membrane components with endoglycosidase F (Endo F) sharpened the diffuse band, carrying Fya activity, obtained by immunoblotting to a band of molecular weight 26 kDa; it was concluded that the Fya protein is heavily glycosylated, with 40–50% of its mass consisting of N-glycan (Tanner et al. 1988).
Fya and Fyb tend to elute from red cells stored in a low pH low-ionic-strength medium, leading to the appearance of substances with specific inhibitory activity for anti-Fya or anti-Fyb in the supernatant fluid. Duffy antigens also elute from red cells after prolonged storage, or mixing, in saline at pH 7.0 (Williams et al. 1981; Mangalmurti et al. 2009).
The structure of Fy glycoprotein (seven membrane spans) is characteristic of the G protein-coupled receptor family which includes chemokine receptors, and the Fy glycoprotein is a promiscuous chemokine receptor, binding to proinflammatory chemokines of both the CXC and CC subfamilies but not homeostatic chemokines (Gardner et al. 2004). However, it lacks the intracellular G protein binding domain (Murphy 1994). Fy glycoprotein is a receptor for IL-8 and melanoma growth stimulatory activity (MGSA); these two chemokines block the invasion of red cells by Plasmodium knowlesi (Horuk et al. 1993). IL-8 binding involves residues on extracellular domains 1 and 4 and requires the presence of intact intrachain disulphide bonds (Tournamille et al. 2003). The Fy glycoprotein is expressed on capillaries and post-capillary venular endothelial cells as well as red cells, and it has been suggested that the Fy glycoprotein has a role in enhancing leucocyte recruitment to sites of inflammation by transporting chemokines across the endothelium (Lee et al. 2003; Pruenster et al. 2009). It seems likely that Fy glycoprotein on red cells and endothelial cells regulate chemokine bioavailability between the circulation and extravascular sites during inflammation (Lee et al. 2006). Since such a regulatory process would be altered in individuals with the Fy(a− b−) phenotype and many patients with sickle cell disease have the Fy(a− b−) phenotype and elicit markers of inflammation, these data raise the possibility of a link between the Fy(a− b−) phenotype and the propensity for alloimmunization upon transfusion of sickle cell patients (reviewed in Anstee 2009 and Anstee 2010). Sickle cell patients with the Fy(a− b−) phenotype are more susceptible to chronic organ damage and proteinuria than patients expressing Fy glycoprotein on their red cells (Afenyi-Annan et al. 2008).
Individuals with the Fy(a− b−) phenotype are more susceptible to asthma (Vergara et al. 2008).
Fy Antigens on Other Cells
Using a sensitive radioimmunoassay, Fy antigens were shown to be absent from platelets (Dunstan et al. 1984). Using immunofluorescence flow cytometry, Fya, Fyb and Fy5 were not detected on lymphocytes, monocytes or granulocytes (Dunstan 1986).
Duffy Groups and Malaria
Fy(a− b−) subjects are protected from infection by Plasmodium vivax (Miller et al. 1976). In parts of Africa, the frequency of Fy is 1.0 and in these areas P. vivax malaria is absent (Welch et al. 1977). The Fy(a− b−) phenotype in these individuals results from a mutation in the binding site for GATA-1, which is upstream of the Fy gene. GATA-1 is a transcription factor that regulates the expression of erythroid genes. The mutation in Fy(a− b−) prevents GATA-1 binding so that the Fy gene is not transcribed and Fy glycoprotein is not expressed in the erythroid cells. Expression in non-erythroid cells is not affected (Tournamille et al. 1995). In Africa, the Fy(a− b−) phenotype occurs on an Fyb genetic background. In Papua New Guinea, the Fy null allele occurs on an Fya genetic background (Zimmerman et al. 1999). The structure of the Fy glycoprotein binding site on Plasmodium knowlesi has been elucidated. The critical binding residues are conserved in P. vivax. Sulphation of Tyr 41 on the Fy glycoprotein increases the affinity of binding by up to 1000-fold suggesting this residue is a critical component of the interaction (Singh et al. 2006).
For P. falciparum infection, there is no association with Duffy groups. The specific ligands concerned in binding the parasites to red cells are the glycophorins (A, B, C and D) and especially the N-acetylneuraminic acid linked in an α2,3 configuration on them (Pasvol et al. 1993; Maier et al. 2003; Tolia et al. 2005; Mayer et al. 2009).
Duffy Groups and HIV
Lachgar and co-worker (1998) report that Duffy glycoprotein on red cells binds HIV and that this binding is inhibited by the chemokine RANTES, which binds the Duffy glycoprotein, but not by MIP-1-α, which does not bind Duffy glycoprotein. They suggest that red cells may be a reservoir for HIV and transmit the virus to leucocytes. Individuals with the Fy(a− b−) phenotype are reported to be more likely to acquire HIV than individuals expressing Fy glycoprotein on their red cells, but once acquired the disease has a slower progression (He et al. 2008). However, these conclusions have been questioned by others (Walley et al. 2009).
Antibodies of the Duffy System
Anti-Fya
This antibody is found mainly following transfusion and, much less commonly, following pregnancy; it is almost never naturally occurring. Anti-Fya occurs about three times less frequently than anti-K (see Table 3.6). Most examples are IgG; about 50% activate complement up to the C3 stage.
Anti-Fya (and -Fyb) are predominantly IgG1; in three series they were solely of this subclass in six out of seven, 12 out of 14 and 11 out of 16 cases respectively (Engelfriet 1978; Hardman and Beck 1981; Szymanski et al. 1982). In the last of these series, three anti-Fya had an IgG2 component and four were partly IgM; two anti-Fyb were entirely IgG1.
Occasionally, anti-Fya agglutinates Fy(a+) red cells suspended in saline and may react with Fy(a+ b−) cells more strongly than with Fy(a+ b+) cells (Race et al. 1953). Some anti-Fya fail to react in manual polybrene and polybrene antiglobulin tests (Malde et al. 1986).
Fya is significantly more immunogenic in Fy(a− b+) white people than it is in Fy(a− b−) black people. In two series, of 25 and 130 patients with anti-Fya, four and 11, respectively, were Fy(a− b−) black people (Kosanke 1983; Vengelen-Tyler 1983); in the second series, none of 11 patients with anti-Fyb was black. All of 29 white people producing anti-Fya were HLA DRB1*04 (Noizat-Pirenne et al. 2006).
Anti-Fyb
Anti-Fyb is about 20 times less common than anti-Fya (Marsh 1975); it is usually found, not on its own, but in sera containing other red cell alloantibodies. A few examples agglutinate red cells suspended in saline. An example of naturally occurring anti-Fyb reacting by the indirect antiglobulin technique has been described by Issitt (1985).
Anti-Fy3, -Fy4 and -Fy5
Anti-Fy3 reacts with all human red cells except those of the phenotype Fy(a− b−). As the latter phenotype is enormously commoner in black people than in white people, it was somewhat unexpected that the antibody is about as rare in black people as in white people. This is probably because the common mutation giving rise to the Fy(a− b−) phenotype in black people (a GATA-1 site mutation, see above) does not affect Duffy glycoprotein expression in non-erythroid tissues, whereas in other races the Fy(a− b−) phenotype has generally had a different genetic background involving inactivating mutations in the Duffy gene itself (Mallinson et al. 1995; Rios et al. 2000). It should be noted that the GATA-1 mutation has also been found in white people (Rios et al. 2000; Pisacka et al. 2001).
The only example of anti-Fy4 reacted with all Fy(a− b−) cells from black people and with most Fy(a+ b−) and Fy(a− b+) cells from black people, i.e. of presumed genotypes FyaFy and FybFy respectively (Behzad et al. 1973). Anti-Fy5 is made by black patients and is often found in sera that contain other immune antibodies; it reacts with most red cells except with Fy(a− b−) cells from black people, with Rhnull cells or with cells carrying variant forms of the e antigen regardless of their Fy status (Meredith 1985).
Clinical Aspects
Duffy antibodies seldom cause severe haemolytic transfusion reactions and only occasionally cause HDN, which is then usually mild (Goodrick et al. 1997). They have been implicated in delayed haemolytic transfusion reactions in multi-transfused black patients with sickle cell disease. Of five patients with anti-Fy3 or anti-Fy5, four had evidence of DHTRs; anti-Fya preceded the appearance of anti-Fy3 or anti-Fy5 (Vengelen-Tyler 1985). In another series, five Fy(a−b−3−) patients with sickle cell disease experienced DHTRs or failed to show the desired HbA increment after transfusion; they all had anti-Fya and other alloantibodies in the serum (Le Pennec et al. 1987).
The development of allo-anti-Fya (and anti-Jkb) 14 months after an allogenic bone graft, which had previously been frozen, has been described in a patient who had not been transfused and not been pregnant (Cheek et al. 1995).
Mimicking anti-Fyb has been found in autoimmune haemolytic anaemia (see Chapter 7). Renal allografts mismatched for Fy antigens have more chronic lesions than matched grafts suggesting that Fy antigens are minor histocompatibility antigens (Lerut et al. 2007). The Fy(a−b−) phenotype is associated with worse clinical outcomes in African Americans with acute lung injury. This may be related to elevated levels of IL-8 in the circulation (Kangelaris et al. 2012).
The Kidd (Jk) System
About 76% of white people possess the Jka antigen; 26% have the genotype JkaJka, phenotype Jk(a+ b−), and 50% have the genotype JkaJkb, phenotype Jk(a+ b+). The remaining 24% have the genotype JkbJkb, phenotype Jk(a− b+). Gene frequencies in white people: Jka, 0.514; Jkb, 0.486 (Race and Sanger 1975). The frequency of Jka is as high as 0.75 in parts of Africa and as low as about 0.25 in some Chinese and Japanese populations (for references, see Daniels 2002).
The type Jk(a− b−) has been found in Filipinos, South-East Asians, Chinese and others and is least uncommon in Polynesians, in whom the frequency is about 1%; it is extremely rare in white people. Two different genetic backgrounds seem to be responsible for the Jk(a− b−) phenotype: a silent recessive gene Jk (Race and Sanger 1975) or a dominant inhibitor gene, In(Jk), which depresses the expression of Kidd antigens (Okubo et al. 1986). Whereas the recessive type completely lacks Jka, Jkb and also Jk3, an antigen which is found in all Jk(a+) and Jk(b+) subjects, traces of these three antigens can be demonstrated in subjects of the inhibitor gene type (Okubo et al. 1986).
Number of Jka and Jkb Sites and Nature of Kidd Protein
The number of Jka sites on JkaJka red cells has been estimated to be 14 000 per red cell (Masouredis et al. 1980).
Red cells carrying Jka or Jkb, when exposed to 2 mol/l of urea in water, rapidly become swollen and spherocytic before lysing, but Jk(a− b−) red cells exhibit shrinkage and crenation and lyse relatively slowly (Heaton and McLoughlin 1982). This behaviour relates to the fact that Kidd antigens are located on a urea transporter protein. It has been estimated that the passage of urea across the red cell membrane is 1000 times slower in Jk(a− b−) red cells than in normal cells (Fröhlich et al. 1991). Individuals with the Jk(a− b−) phenotype have a urine-concentrating defect (Sands et al. 1992).
The Kidd glycoprotein (synonym: HUT-B1) comprises 389 amino acids and has 10 membrane-spanning domains with its amino and carboxyl termini in the cytoplasm. A single extracellular N-glycosylation site is located at Asn211 on the third predicted extracellular loop. The Jka/Jkb polymorphism results from an Asp280Asn transition located in the fourth predicted extracellular loop of the protein (Lucien et al. 2002). The Jk(a− b−) phenotype can result from a variety of different mutations in the gene (reviewed in Daniels 2002; see also Wester et al. 2008; Liu et al. 2009).
Kidd antigens were found to be absent from platelets when using a sensitive radioimmunoassay (Dunstan et al. 1984) and from lymphocytes, monocytes and granulocytes when using immunofluorescence flow cytometry (Dunstan 1986).
Antibodies
The first example of anti-Jka was discovered in the serum of a woman who had given birth to an infant with HDN (Allen et al. 1951). Anti-Jkb is rarer than anti-Jka, and is usually found in sera that contain other immune blood group antibodies. The antibody anti-JkaJkb (i.e. inseparable), also known as anti-Jk3, was present in the serum of the first Jk(a− b−) patient described (Pinkerton et al. 1959).
Anti-Jka and anti-Jkb occur only as immune antibodies; they seldom agglutinate saline-suspended red cells and are mainly IgG but may be at least partly IgM (Polley et al. 1962). Although the paper just quoted reported that all of 15 examples of anti-Jka bound complement, a later study of a larger number of Kidd antibodies (55 anti-Jka and 15 anti-Jkb) showed that only about 50% were complement-binding (G Garratty, personal communication). Yates and co-workers (1998) report that Kidd antisera bind complement only when at least a trace of IgM antibody is present and that serum fractions containing only IgG antibody do not bind complement. This observation may explain the fact that the pattern of destruction of incompatible red cells by Kidd antibodies is often characteristic of that produced by non-complement-binding antibodies (see Chapter 10). Anti-Jka reacts far more strongly with JkaJka than with JkaJkb cells and may be undetectable with the latter.
Anti-Jka and anti-Jkb were found to be predominantly IgG3 in three out of three cases (Engelfriet 1978). In another series, of 17 cases, most antibodies were a mixture of IgG1 and IgG3 but some were partly IgG2, some partly IgG4 and some partly IgM (Hardman and Beck 1981).
Three IgM human monoclonal anti-Jkb and one IgM anti-Jka have been produced. The antibodies agglutinate red cells, even of phenotype Jk(a+ b+), in saline after an immediate spin (Thompson et al. 1991).
Jk(a−) individuals who have made anti-Jka are likely to express HLA-DRB1*0101, DRB1*0102 or DRB1*1001 (Reviron et al. 2005).
Clinical Aspects
Both anti-Jka and anti-Jkb can cause haemolytic transfusion reactions. Kidd antibodies are particularly associated with DHTR, being responsible for about one-third of all cases (see Chapter 11). This association is due, at least in part, to the fact that it is not uncommon for Kidd antibodies to become undetectable within a few months of first being detected. Kidd antibodies only rarely cause HDN, a fact for which there is no obvious explanation.
Anti-Jka has occasionally been found as an autoantibody in autoimmune haemolytic anaemia (see Chapter 7). A transient auto-anti-Jkb was apparently stimulated by proteus urinary tract infection; Jk(b−) red cells pre-incubated with Proteus mirabilis became agglutinable by anti-Jkb (McGinniss et al. 1979).
Temporary suppression of Jka was observed in a patient of phenotype Jk(a+ b−), in association with the development of anti-JkaJkb (anti-Jk3); the antibody appeared to be responsible for a haemolytic reaction following the transfusion of Jk(a+ b−) blood (Issitt et al. 1990). Renal allografts mismatched for Kidd antigens had more interstitial inflammation than matched grafts suggesting Kidd antigens may be minor histocompatibility antigens (Lerut et al. 2007).
The MNSs System
Antigens
The antigens M and N were discovered by injecting human red cells into rabbits, absorbing the resulting immune serum with one sample of human cells and showing that it would still react with other samples (Landsteiner and Levine 1927).
M and N are alleles giving rise to three genotypes, MM, MN and NN, with frequencies in white people of about 28%, 50% and 22% respectively. The corresponding phenotypes are written M+ N−, M+ N+ and M− N+. When tested with most anti-M, M+ N− red cells react more strongly than M+ N+ cells. Red cells of the phenotype M+ N− show some reaction with anti-N except when they come from a subject who is S negative, s negative, because the Ss glycoprotein (syn. glycophorin B) also carries an amino-terminal determinant with N specificity, sometimes written ‘N’ (see below).
As described in more detail below, in the section on the chemistry of MN and Ss blood group systems, M and N are carried on glycophorin A (GPA, MN glycoprotein) and the antigens S and s are carried on glycophorin B (GPB, Ss glycoprotein). The inheritance of S and s is closely associated with that of M and N, i.e. the Ss locus is very close to the MN locus. The antigen S is found about twice as frequently in MM as in NN subjects. Among white people, about 55% are S positive and 89% are s positive (Race and Sanger 1975). Haplotype frequencies in white people are approximately as follows: MS, 0.247; Ms, 0.283; NS, 0.080; Ns, 0.390. About 1.5% of black people are S− s− but this phenotype has not been found in white people.
Rare Genetic Variants Associated with MN and Ss
There are many rare genetic variants associated with the MNSs blood group system. Some are due to absence of glycophorin A (GPA) or glycophorin B (GPB) or both, namely (1) complete absence of GPA, as in some types of En(a−) red cells; (2) complete absence of GPA and GPB, as in the extremely rare genotype MkMk; and (3) complete absence of GPB, as in subjects with red cells of the phenotype S− s−.
Numerous low-frequency antigens have been identified; some of these result from amino acid substitutions or glycosylation changes, or both, in GPA or GPB; most are associated with abnormal hybrid glycophorin molecules (partly GPA and partly GPB: for reviews, see Issitt and Anstee 1998; Daniels 2002). It has been suggested that the high rate of structural change in the glycophorins provides a means of evasion of P. falciparum (Wang et al. 2003) or that the variant glycophorins act as more general decoy receptors attracting pathogens to the anucleated red cell and away from their target tissues (Baum et al. 2002).
En(a−)
There are at least two kinds of En(a−) red cells: (1) total absence of GPA but a normal GPB and therefore some trypsin-resistant ‘N’ activity (Furuhjelm et al. 1969); and (2) total absence of N and ‘N’ but a weak trypsin-resistant M (Darnborough et al. 1969); it has been shown that this represents a hybrid molecule of a normal M-active GPA aminoterminal segment with the carboxy-terminal segment from a normal S-active GPB, i.e. a GP(A-B) hybrid (Dahr et al. 1978).
Both kinds of En(a−) red cells have a greatly reduced sialic acid content and electrophoretic mobility. The red cells (if Rh D positive) are agglutinated in saline by IgG anti-D.
Sialic acid deficiency per se does not seem to affect red cell survival, as the En(a−) condition is not associated with a haemolytic anaemia. Moreover, Tn red cells, which are also sialic acid deficient, may survive normally (see Chapter 7). On the other hand, sialic acid deficiency of red cells induced experimentally with neuraminidase in animals is associated with gross shortening of red cell survival (Jancik and Schauer 1974; Durocher et al. 1975).
The use of the terms Ena antigen and anti-Ena is inappropriate, as what is being observed is the inherited deficiency of GPA and the antibody response of GPA-deficient individuals to the surface of normal red cells (Anstee 1981).
Mk is regarded as a silent allele resulting, in homozygotes, with the absence of MN and Ss antigens. Two MkMk individuals had red cells which completely lacked the GPA and GPB and had a sialic acid content of between 25% and 31% of normal; both subjects were normal haematologically (Tokunaga et al. 1979). Very few other MkMk propositi have been found but Mk heterozygotes, whose red cells, such as those of En(a−) heterozygotes, have reduced sialic acid levels, are commoner (approximate frequency of Mk, 0.0005). For references, see Daniels (2002). GPA associates with the anion transport protein band 3 in the red cell membrane (see also Diego blood groups in this chapter) and absence of GPA in En(a−) and MkMk red cells results in band 3 adopting a structure with lower anion transport activity than when GPA is present (Bruce et al. 2003).
S− s−
This phenotype occurs mainly in black people; it has been found in one Indian family but not yet in white people. S− s− red cells lack ‘N’ and M+ N− S− s red cells are thus the only ones that are completely N negative. S− s− American black people either completely lack the U antigen or have a weak variant form of it (Issitt and Anstee 1998).
U
The U antigen is found in all white people and in about 99% of black people. U-negative subjects are always S− s−.
In some Rhnull cells the Ss and U antigens may be difficult to detect, especially when using the IAT. The amount of GPB was found to be reduced to one-third in Rhnull cells. It is believed that, during biosynthesis, Rh proteins form a complex with GPB facilitating its incorporation into the red cell membrane (Dahr et al. 1987; see also Chapter 5).
The Miltenberger (Mi.) Series of Antigens
These are all carried on glycophorins A or B on hybrid molecules composed of both glycophorins. It has been proposed that these antigens, which formerly had a wide variety of names (Vw, Hut, Mur, etc.) and were also termed Mi.I–XI, should no longer be regarded as belonging to a subsystem of MNSs. Instead they should be termed GP. Vw, GP. Hut, etc. (GP standing for glycophorin). For a fuller description, see Tippett and co-workers (1992); for a review of the chemistry of the antigens, see Dahr (1992) and Daniels (2002). The existence of antibodies of specificity anti-Mia has been questioned but such a specificity has been reported for a monoclonal antibody (Chen et al. 2001).
In white people, the frequency of each of the various Mi antigens is less than 1 per 1000 but the Mi.III phenotype (predominantly GP.Mur) is relatively common in South-East Asia, with a frequency reaching 10% in Thailand, 7% in Chinese and 88% in the Ami mountain people of Taiwan (Broadberry and Lin 1996). Anti-‘Mia’, that is to say an antibody reacting with Mi.III red cells but not tested with other Mi cells (probably anti-Mur), has a frequency of 0.28% in Chinese patients in Taiwan and is, in that region, the commonest alloantibody of clinical significance. It may be responsible for both HDN and, in all probability, HTR (Broadberry and Lin 1994; Lin and Broadberry 1994). The high frequency of this antibody in Asian populations necessitates the inclusion of Mur-positive red cells in antibody screening panels. GP.Mur is an altered GPB molecule which results from splicing a small segment of exon 3 of the GPA gene into the GPB gene to create a GP(B – A – B) hybrid protein with residues 1–48 from GPB, residues 49–57 from GPA and residues 58–103 from GPB (Huang and Blumenfeld 1991). The resultant hybrid glycoprotein is more heavily sialylated than GPB (Anstee et al. 1979). Human IgM monoclonal anti-Mur has been produced (Uchikawa et al. 2000).
Numbers of S, s and U Antigen Sites
The number of available s sites on ss red cells was found to be approximately 12 000 and of available U sites on U-positive red cells to be about 17 000 (Masouredis et al. 1980). These numbers are clearly far less than the number of potential sites; it has been estimated that each red cell has about 2.5 × 105 copies of GPB and, incidentally, about 1 × 106 copies of GPA (see review by Anstee et al. 1982).
MNSs Antigens on Other Cells
M, N, S, s and U antigens were not detected on lymphocytes, monocytes or granulocytes by immunofluorescence flow cytometry (Dunstan 1986). Using a sensitive radioimmunoassay, MNSs antigens could not be detected on platelets (Simpson et al. 1987).
Chemistry of the MN and Ss Antigens
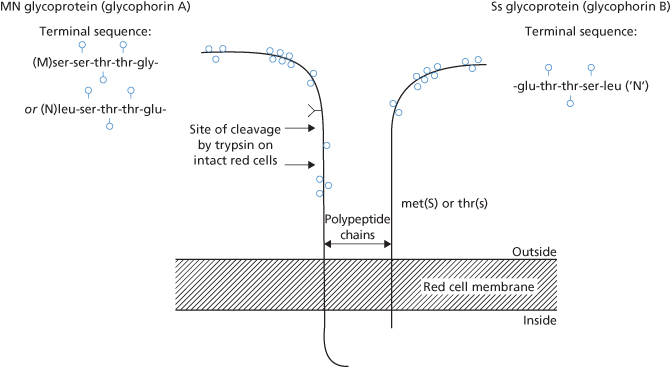
As described above, MN and Ss antigens are found respectively on glycophorins A and B of the red cell membrane (Figure 6.1).
Figure 6.1 The MN and Ss glycoproteins. s, O-linked glycans; Y, single N-linked glycan on GPA.
(Source: Anstee 1980. Reproduced with permission of John Wiley & Sons Ltd.)
The glycoproteins of the red cell membrane can be separated by SDS-PAGE (see Chapter 3); glycoproteins rich in sialic acid (glycophorins) can be stained with PAS (periodic–acid Schiff).
The only difference between M and N is that the polypeptide chain determining M has the aminoterminal sequence ser-ser–thr–thr–gly– and that the chain determining N has the amino-terminal sequence: leu–ser–thr–thr-glu–. Some examples of monoclonal anti-M react with the ‘ser’ region of the determinant and others with the ‘gly’ region (Nichols et al. 1985).
S and s are carried on glycophorin B and differ from one another in a single amino acid substitution, i.e. at position 29 there is a methionine in S and a threonine in s (Dahr et al. 1980a,b). The Ss glycoprotein on intact red cells is not affected by trypsin treatment but is inactivated by pronase, by high concentrations of chymotrypsin (Judson and Anstee 1977) and by bromelin, ficin and papain.
The first 26 residues of the Ss and the N-specific MN glycoproteins are identical (Dahr et al. 1980a,b), thus explaining the fact that red cells of M+ subjects, unless they are S− s−, react with anti-N. Nevertheless, with anti-N the reactions of ‘N’ (see above) and N are not identical: with anti-N, MM red cells react less strongly than MN red cells, which, in turn, react less strongly than NN red cells.
It seems that oligosaccharides (sialic acid residues) attached to terminal amino acids of the MN glycoprotein, as well as the amino acids themselves, are involved in M and N specificity (Uhlenbruck et al. 1976; see also Judd et al. 1979).
Antibodies of the MNSs System
Anti-M
In adults, anti-M is a relatively common naturally occurring antibody reacting optimally at 4°C; it usually reacts weakly or not at all at 37°C. In blood donors, the prevalence of naturally occurring anti-M, detectable in microplates with saline-suspended cells at room temperature, is 1 in 2500 with M+ N− cells, and 1 in 5000 with M+ N+ cells (A Lubenko, personal communication). A higher incidence has been found using an AutoAnalyzer and a low-ionic-strength polybrene method (Perrault 1973).
Anti-M is distinctly commoner in infants than in adults (Strahl et al. 1955). In infants in hospital in Toronto, approximately 4% of agglutinins active at room temperature were anti-M, whereas in adults the figure was approximately 1% (personal communication reported in previous editions of this book).
The development of anti-M in Rh D-immunized subjects following a series of boosting injections has been reported (Wiener 1950; Teesdale et al. 1991). In the latter publication, anti-M developed in 3 out of 25 NN subjects, but in two of the three the antibody was very weak and reactive only at room temperature.
Although almost all examples of anti-M occur in M− N+ subjects, a few have been found in M+ N+ subjects, although not as autoantibodies (e.g. Konugres et al. 1966). Anti-M occurring as an autoantibody in M+ N+ subjects has also been described (e.g. Fletcher and Zmijewski 1970).
Anti-M Detectable Only at Reduced pH
Some examples of anti-M can be detected only after the serum has been acidified. Twenty-one such examples were found in testing the plasma of 1000 group N donors (Beattie and Zuelzer 1965). The presence of anti-M should be suspected when an agglutinin is found in the plasma of an ACD (or CPD) sample from a donor whose unacidified serum has no abnormal antibodies.
Anti-M Reacting Only with Glucose-Treated Red Cells
This was described by Reid and colleagues (1981). The antibody agglutinated M-positive red cells that had been incubated in 2% glucose for a minimum of 2 h at 37°C or for substantially longer periods at low temperatures. It also agglutinated red cells from patients with diabetes mellitus. Red cells that had become agglutinable following glycosylation could be rendered unagglutinable by incubation in saline. The anti-M activity was totally inhibited by adding an equal volume of 1% glucose to the serum. Fourteen other examples of human anti-M were not inhibited by glucose. Another anti-M agglutinin reacted with red cells incubated with either glucose or galactose and was inhibited by 2% glucose, mannose or maltose (Morel et al. 1981).
Anti-M1 is found as an additional specificity in about one in three sera containing anti-M; it has only rarely been found on its own. M1 is commoner in black than in white people (Race and Sanger 1975).
Immunoglobulin Class of Anti-M
Of 10 examples of human anti-M, all were at least partly IgM (P Rubinstein personal communication), although naturally occurring anti-M may be solely IgG (Mollison 1972, fifth edn, p. 332). In a series of 45 cases, anti-M was at least partly IgG in 34, being solely IgM in the remainder (Smith and Beck 1977).
Most anti-M react more strongly in albumin than in saline: one example associated with HDN reacted as well at 37°C as at 4°C (Stone and Marsh 1959) and was subsequently shown to be IgG (Adinolfi et al. 1962).
Apart from one case of allo-anti-M (see Chapter 10) and one of auto-anti-N (see below), no examples of anti-M or anti-N have been shown to bind complement using ordinary serological methods. However, in an investigation with 125I-labelled anti-Cad, a small amount of complement appeared to be bound by one example of IgM anti-M; two examples of IgG anti-M did not bind complement (Freedman et al. 1980).
Murine monoclonal antibodies to M, N and s are available. Two examples of anti-M were shown to react with different epitopes (see above); at room temperature, the antibodies had titres of 128–256 with M+ N− red cells and of eight with M− N+ cells (Nichols et al. 1985).
The reactions of monoclonal anti-M and anti-N, such as those of some polyclonal anti-M (see above), are greatly affected by pH. Testing the pH dependency of MN monoclonals is important because, in some instances, the specificity of agglutination reactions can be greatly improved by selection of the proper pH (Lisowska 1987). Five monoclonal anti-s were produced using a synthetic peptide corresponding to residues 17–36 of glycophorin B encompassing Thr29, the residue critical for s antigen activity (Halverson et al. 2009).
Other Non-Human Sources of Anti-M
Good anti-M sera can be produced in rabbits (see Menolasino et al. 1954). The anti-M lectin prepared from the seeds of Iberis amara (candytuft) does not seem to be a useful reagent.
Allo-Anti-N
Anti-N is rarer than anti-M; in a series of 86 000 (excluding patients on renal dialysis, see below), only two examples were found (B Croucher, personal communication).
Although naturally occurring, anti-N is typically an IgM cold agglutinin, inactive above a temperature of 20–25°C; one naturally occurring IgG anti-N has been encountered (ER Giblett, personal communication). It is very rare for anti-N to be formed as an immune antibody. The first example was described by Callender and Race (1946). In another case, IgG anti-N developed after multiple transfusions in a subject of phenotype M+ N− S− s− U− (Ballas et al. 1985).
The demonstration that the Ss glycoprotein carries ‘N’ antigen (see above) explains the fact that anti-N agglutinates M+ cells and also agglutinates trypsin-treated M+ cells, as the ‘N’ antigen on Ss glycoprotein is not destroyed by trypsin. As expected, anti-N does not react with M+ N−, S− s− red cells. Similarly, potent anti-N is found almost exclusively in S− s− subjects (Telischi et al. 1976; see also above).
Anti-N reacting only with glucose-treated red cells was found in three different sera (Morel et al. 1975). The reactions were inhibited by adding glucose to the serum. Glucose-dependent anti-N requires a determinant resulting from the reaction of glucose with the N-terminal amino acid of glycophorin A, but no sialic acid, for binding (Dahr et al. 1981; Morel et al. 1981).
Cold-Reacting Auto-Anti-N
In screening 45 000 blood samples against M− N+ (or M+ N+) cells in the AutoAnalyzer, using a low-ionic-strength polybrene method, low-titre anti-N not detectable by manual methods was found in six M+ N+ subjects and in one M+ N subject; a more potent example was found in one M− N+ subject (Perrault 1973). The last example was remarkable in that it was IgG and could be detected by manual methods only by using a 10–20% cell suspension, no agglutination being obtained with a 2% cell suspension. The antibody was active at 4°C but not at 12°C or at higher temperatures.
Anti-Nf Developing in Patients on Chronic Haemodialysis
Patients undergoing regular haemodialysis, irrespective of their MN phenotype, may develop anti-N, usually active at 20°C as well as 4°C, but never active at 37°C (Howell and Perkins 1972). The stimulus for anti-N production comes from small amounts of red cells left in dialysis equipment previously sterilized by formaldehyde treatment: an antigen ‘Nf’ develops. Exposure of M+ N− red cells to 0.06% formaldehyde induces Nf specificity after only 30 s at 37°C; at room temperature exposure to a concentration as low as 0.002% is sufficient to induce Nf specificity in 20 h (Gorst et al. 1977). In one series, anti-Nf was found in 68 out of 325 patients on renal dialysis, all of whom had been treated with a formaldehyde-sterilized dialyser (Fassbinder et al. 1978).
Although anti-Nf, being inactive at 37°C, is not expected to destroy transfused red cells, a case has been described in which the antibody appeared to be responsible for the rejection of a chilled transplanted kidney. When a second kidney from the same cadaver was perfused with warm saline before transplantation, grafting was successful (Belzer et al. 1971).
Anti-N Lectins
Anti-N is present in the seeds of Vicia graminea and of Bauhinea purpurea var. alba. Of the two, the lectin of V. graminea is the more potent. The anti-N lectin from V. graminea reacts not only with N+ red cells but also with neuraminidase-treated red cells of all MN phenotypes; the receptor with which the lectin reacts is known as Nvg.
The lectin from Moluccella laevis carries anti-A and anti-N on the same molecule (Bird and Wingham 1970). The lectin also reacts strongly with T and Tn cells (Bird 1978).
Anti-N lectin from V. graminea reacts with both normal N and ‘formaldehyde N’ red cells but the lectin from M. laevis reacts only with normal N (of group B or O subjects) (Bird and Wingham 1977).
Anti-S, Anti-s, Anti-U
Anti-S has occasionally been found as a naturally occurring antibody, but is more commonly found as an immune antibody in patients who have received many transfusions.
Some examples of ‘incomplete’ anti-S can be detected much more readily by incubating serum and red cells at room temperature rather than at 37°C (Lalezari et al. 1973, see below). Although most examples of ‘immune’ anti-S are incomplete IgG antibodies, two examples behaving as saline agglutinins were found to be IgM (Adinolfi et al. 1962).
Antibodies to low-frequency antigens are commonly found in anti-S sera; of nine anti-S investigated with a collection of S-negative cells with rare phenotypes, seven were found to react with one or more cells and two sera contained 15 specificities each (A Lubenko and M Contreras, personal observations).
Anti-s is rare; it may be IgG or IgM.
In tests in an AutoAnalyzer, all of seven examples of anti-s and one of anti-U (all IgG) reacted more strongly at low temperatures. Similarly, using the IAT, three examples of anti-s and one of anti-S gave stronger reactions at low temperatures, whereas examples of anti-D, -K and -Fya reacted more strongly at 37°C (see also Chapter 8).
Anti-U is also rare; as a rule it is a non-complement-binding IgG antibody (Issitt 1981). The antibody is found exclusively in black people. There are two kinds of anti-U, one reacting with about 1.25% of black people and one with about 0.25%, i.e. 1% of black people have a U variant antigen (see Issitt 1990). Anti-U is occasionally encountered in frequently transfused patients with sickle cell disease.
Anti-S and anti-U were found to be IgG1 with or without other subclasses in 13 out of 17 examples tested. On the other hand, anti-s was found to be solely IgG3 in four out of five cases (Hardman and Beck 1981).
Other Antibodies of the MNSs System
Anti-Vw and anti-Mg have frequencies of 1–2% and most commonly occur as agglutinins reacting more strongly at room temperature than at 37°C. Anti-Vw, which reacts only with Miltenberger class I cells (MLI), occasionally develops immune characteristics and its specificity then broadens to become anti-Mia (reacting with Mi.I, II, III, IV and VI cells) (TE Cleghorn, personal communication).
As mentioned above, in Taiwan the frequency of Mi.III was found to be 7.3% and ‘anti-Mia’, i.e. an antibody reacting with Mi.III red cells but not tested with other Mi antigens, was found to be the most frequently occurring immune alloantibody (Broadberry and Lin 1994). HLA-DRB1*0901 is more common in individuals making ‘anti-Mia’ (Chu et al. 2009).
‘Anti-Ena’
As mentioned above, there is no such antigen as Ena, although subjects who lack glycophorin A [‘En (a−)’] may make a spectrum of antibodies, ‘anti-Ena’, which are directed against different portions of the glycophorin A molecule (Pavone et al. 1981; Vengelen-Tyler et al. 1981). Anti-Ena has only once been found as a naturally occurring antibody (Taliano et al. 1980), the other examples having been found following transfusion or pregnancy.
The term ‘auto-anti-Ena’ has been used to describe a heterogeneous group of related but not identical specificities, several of which recognize different portions of the MN sialoglycoprotein (Pavone et al. 1981). Auto-anti-Ena is found in certain patients with the warm antibody type of autoimmune haemolytic anaemia.
Clinical Aspects
Most antibodies of the MNSs system are not active at 37°C and are thus of no clinical significance. Isolated examples of wider thermal amplitude have been shown to destroy test doses of incompatible red cells or cause HTRs, e.g. anti-M (Wiener 1950; see also Chapters 10 and 11); anti-Mia (Cutbush and Mollison 1958); anti-N (Ballas et al. 1985); anti-S (Cutbush and Mollison 1949) and anti-s (Fudenberg and Allen 1957; see also Chapter 10). Anti-U can cause immediate haemolytic transfusion reaction (IHTR) (Wiener et al. 1953) and DHTR, particularly in frequently transfused patients with sickle cell disease (Davies et al. 1986). HDN caused by MNSs antibodies is described in Chapter 12.
Antibodies to low-frequency antigens such as Vw are of negligible importance in blood transfusion because of the rarity of the corresponding antigen. One of the very few haemolytic transfusion reactions associated with antibodies to low-frequency antigens occurred in a patient who happened to be transfused more than once with blood from the same donor: a carrier of the low-frequency antigen Kamhuber (Speiser et al. 1966), later shown to be identical with Far (Giles 1977). Some examples of antibodies to the low-frequency antigens of the MNSs system, for example anti-Mg, -Vw, -Ria, -Dantu, -Hop and -Nob, have been naturally occurring; other examples, such as anti-Vw, Mta, Mit and -Mv, have been discovered in the serum of women whose infants have been affected with HDN. Whereas in blood transfusion the chance of receiving more than one unit carrying one of these antigens is very low when random blood donors are used, in women who have several children by a man who carries one of the antigens, the chance of allo-immunization is obviously far higher.
Mi.III is not a low-frequency antigen in Chinese in Taiwan and, there, a corresponding antibody is the commonest alloantibody encountered in patients (Broadberry and Lin 1994).
Cold-reacting anti-N and warm-reacting anti-U have been found as autoantibodies (see Chapter 7).
Glycophorin A Is a Receptor for C3b
Venom from the Loxosceles spider induces intravascular haemolysis by rendering red cells susceptible to lysis by human complement. The mechanism by which this occurs appears to involve activation of an endogenous metalloproteinase, which cleaves glycophorins (GPA, B and C) from the red cell surface (Tambourgi et al. 2000, 2002). GPA is an inhibitor of red cell lysis by autologous complement (Okada and Tanaka 1983; Tomita et al. 1993).
Karnchanaphanurach et al. (2009) provide evidence that when complement is activated using cobra venom factor, C3b binds to glycophorin A stimulating the formation of a C3b-glycophorin A-band 3 protein complex and that decay accelerating factor (DAF, syn CD55, see also ‘Cromer’) which also binds C3b, becomes immobilized in this complex. They show that formation of the complex is associated with an increase in membrane stiffness by altering membrane skeletal interactions, and suggest this may provide a mechanism whereby senescent red cells are removed from circulation. Craig et al. (2005) found that when complement was activated with IgM or immune complexes bound to glycophorin A, the IgM-C3b and immune complex −C3b complexes colocalized with CR1 (CR1, see also ‘Knops’) and not with DAF. They consider that immune complexes bound to CR1 and DAF are removed through interaction with Fc receptors on macrophages in liver and spleen by a proteolytic mechanism causing the affected red cells to be released back into the peripheral circulation with reduced levels of CR1 and DAF. Immune complexes bound to glycophorin A were processed in a similar manner to those bound to CR1 and DAF, except for a small proportion of red cells (<15%) which were phagocytosed. Since apparently healthy individuals with a total deficiency of GPA are known (see discussion of En(a−) and MkMk above) it seems likely that other C3b binding processes can substitute for the lack of GPA in these individuals.
Glycophorins A and B Are Receptors for Plasmodium Falciparum and Babesia Divergens
Different strains of the malarial parasite P. falciparum bind to red cells through the sialic acid rich glycophorins A, B or C/D (Pasvol et al. 1993; Maier et al. 2003; Tolia et al. 2005; Mayer et al. 2009). Babesia divergens, a causative agent of babesiosis in man, shows reduced invasion of red cells of phenotype En (a−) and S−s−U− suggesting that GPA and GPB are also receptors for this protozoan parasite ( Lobo 2005).
The Lutheran (Lu) System
The first example of an antibody revealing the existence of this system was found in a transfused patient (Callender et al. 1945).
About 8% of Europeans, Africans and North Americans are Lu(a+), but the antigen is rare or absent in other populations. Almost all Lu(a+) subjects have the genotype LuaLub, the overall frequency of the genotype LuaLua being only 0.15%. Virtually all other subjects (approximately 92% of the population) have the phenotype Lu(a− b+) (gene frequencies: Lua, 0.039; Lub, 0.961). Very rarely, subjects have the phenotype Lu(a− b−) (Crawford et al. 1961); the first example to be detected was found to be due to a dominant inhibitor gene, In (Lu), which is the least uncommon genetic background of Lu(a− b−). In (Lu) also inhibits the production of P1, i, Inb and the high-incidence antigen, AnWj. Thus, antibodies which fail to react with Lu(a− b−) samples may not always belong to the Lutheran system. Subjects with an In (Lu) gene are reported to have some red cell abnormalities. On incubation in plasma for 24 h, osmotic fragility decreases, owing to a reduction in total cation content (Udden et al. 1987). Although survival in vivo of autologous red cells is normal, lysis in vitro after 7 days’ storage in Alsever’s solution was five times that of normal red cells, and the subjects are presumably unsuitable as donors (Ballas et al. 1992). The In(Lu) Lu(a− b−) phenotype results from inheritance of inactivating mutations on one allele of the erythroid transcription factor EKLF (Singleton et al. 2008).
Other examples of the Lu(a− b−) phenotype are due to a double dose of a recessive gene Lu or to an X-borne inhibitor, XS2, of expression of the Lu antigen (see review by Crawford 1988). The Lu(a− b−) phenotype resulting from a double dose of a recessive gene is caused by inactivating mutations in the Lutheran gene (Crew et al. 2007). The Lu(a− b−) phenotype resulting from the X-borne inhibitor gene XS2 is caused by a mutation in the erythroid transcription factor GATA-1 (Ter414Arg). This mutation predicts a translated GATA-1 protein containing an additional 41 amino acids at the carboxy terminus (Singleton et al. 2009a).
With anti-Lub, Lu(a−b+) samples from newborn infants react more weakly than those from adults (Kissmeyer-Nielsen 1960), and Lu(a+ b+) samples from newborn infants react very weakly indeed (Greenwalt et al. 1967).
Transient depression of Lua with the production of an antibody against a high-frequency Lu antigen has been observed in a patient with AITP (see Chapter 7).
In the numbered nomenclature, Lua is Lu1 and Lub is Lu2; Lu3 is an antigen present on all except Lu(a− b−) red cells. There are three pairs of antithetical antigens in addition to Lu1 and Lu2 which have been shown to be inherited at the Lu locus: Lu6 and Lu9; Lu8 and Lu14; Lu18 (Aub) and Lu19 (Aub). Only the latter pair are polymorphic; Lu6 and Lu8 are high-frequency antigens and Lu8 and Lu14, low. In addition to the aforementioned antigens, there are 11 high-frequency antigens (Lu4, Lu5, Lu7, Lu11, Lu12, Lu13, Lu16, Lu17, Lu20,Lu21 and LURC).
Using a sensitive radioimmunoassay, the main Lu antigens could not be detected on platelets (Dunstan et al. 1984) and, using immunofluorescence flow cytometry, Lub could not be detected on lymphocytes, monocytes or granulocytes (Dunstan 1986). On the other hand, the Lutheran glycoprotein is widely expressed in human tissues (Parsons et al. 1995).
Chemistry of Lutheran Antigens
Using a murine monoclonal anti-Lub, two glycoproteins, a major protein of apparent mol. wt. 85 kDa and a minor protein of 78 kDa carrying Lub activity have been identified (Parsons et al. 1987). The 85-kDa form of the Lutheran glycoprotein comprises 597 amino acids with a large amino-terminal extracellular domain of 518 residues, a single membrane traverse and a small cytoplasmic domain of 59 residues (Parsons et al. 1995). The 78-kDa form has an alternatively spliced cytoplasmic domain of 19 residues and corresponds to a protein previously described as B-CAM (Rahuel et al. 1996). The extracellular domain of the Lutheran glycoprotein comprises five immunoglobulin superfamily domains and contains five potential sites for N-glycosylation, one on domain three and four on domain four (Parsons et al. 1995). The polymorphic antigens Lua/Lub and Aua/Aub are located on domains one and five, respectively, and arise from nucleotide substitutions that change a single amino acid (Lua/Lub, His77Arg; Aua/Aub,Thr539Ala: Parsons et al. 1997). Analysis of the location and structure of the other antigens in the Lutheran system has shown there are antigens on four of the five Ig domains and each is defined by nucleotide substitutions that encode single amino acid substitutions (Crew et al. 2003; Colour Plate 6.3).
The 3-dimensional structure of amino terminal domains 1 and 2 of the Lutheran glycoprotein has been determined by X-ray crystallography and that of domains 3, 4 and 5 deduced from small angle X-ray scattering. The glycoprotein forms an extended structure with a distinctive bend between the second and third domains (Mankelow et al. 2007; Colour Plate 6.3).
Using a murine monoclonal anti-Lub the number of Lub sites per red cell was estimated to be about 2000–4000 on LubLub cells and 1000–2000 on LuaLub cells (Merry et al. 1987).
The Lua antigen is destroyed by trypsin, chymotrypsin and pronase (Judson and Anstee 1977). Lutheran antigens are destroyed when red cells are treated with thiol reagents (DTT or AET: Levene et al. 1987), an observation consistent with antigenic structures located on Ig domains whose structures are defined by intrachain disulphide bonding.
Antibodies of the Lutheran System
Anti-Lua
This antibody usually occurs as an agglutinin, reacting more strongly at room temperature than at 37°C. Typically, although large agglutinates are formed, many cells are unagglutinated (Callender and Race 1946). Anti-Lua is most commonly IgM but may be IgG or a mixture of IgM and IgG, and may also be partly IgA. Of 19 examples, only three failed to agglutinate red cells suspended in saline and these three appeared to be solely IgG; of the remaining 16, at least nine were partly IgG and at least two partly IgA, as judged by the fact that agglutinating titres were enhanced by anti-IgG or anti-IgA respectively (A Lubenko, personal communication).
In a deliberate search for anti-Lua, only three examples were found in more than 18 000 donors (Greenwalt and Sasaki 1957). Attempts to stimulate the formation of the antibody by injecting Lu(a+) red cells have given apparently conflicting results. In one series, two out of eight patients developed anti-Lua 2–4 weeks after the transfusion of a single unit of blood, although the antibody was detectable only transiently (Mainwaring and Pickles 1948). In another series, in which four injections of relatively small amounts of blood were given at 3-monthly intervals to 12 subjects, none formed antibody (Race and Sanger 1954, p. 208). Perhaps the apparent discrepancy is explained by the fact that large amounts of red cells are required to induce immunization to Lua.
Anti-Lub
The first example of this antibody was found in an Lu(a+ b−) woman who had never been transfused but had had three previous pregnancies (Cutbush and Chanarin 1956). Anti-Lub is rare, as expected from the rarity of the Lu(a+ b−) phenotype; all examples have been found in patients who have either been pregnant or been transfused.
Most examples of anti-Lub appear to be mixtures of IgM and IgG. In one laboratory, 14 out of 16 examples agglutinated red cells in saline, the remaining two being solely IgG; of the 14 agglutinins all were enhanced by anti-IgG but only four were enhanced by anti-IgA (A Lubenko, personal observations). In another laboratory, all examples were at least partly IgG although some may have been partly IgM; none was IgA (WL Marsh, personal communication).
Of 11 IgG anti-Lub, all were believed to be at least partly IgG4 (Hardman and Beck 1981). However, as most anti-Lub are agglutinins and as IgG subclasses cannot be determined reliably on DTT-treated sera, the finding is difficult to confirm.
Anti-LuaLub(Anti-Lu3)
An antibody with this specificity in which the anti-Lua and anti-Lub activities were inseparable was found by Darnborough and colleagues (1963) in a woman of the rare phenotype Lu(a− b−). The antibody may have been immune as it was absent from the woman’s serum before she had a transfusion 13 years previously and was also absent during a pregnancy that she had borne 6 years previously. This, and subsequently described examples of anti-LuaLub reacted more strongly by IAT than by agglutination tests. The antibody seems to be made by subjects of the recessive type and not by those of the dominant type of Lu(a− b−).
Other Lutheran Antibodies
Many other antibodies against high-frequency antigens, for example anti-Lu4, etc., have been described; all fail to react with Lu(a− b−) red cells but react with the red cells of other rare Lutheran phenotypes (Daniels 2002).
Two monoclonal antibodies detecting high-frequency antigens absent from red cells of the dominant type of Lu(a− b−) were later identified as anti-AnWj (Knowles et al. 1982).
Clinical Aspects
Anti-Lua has not been clearly incriminated as a cause of increased red cell destruction. In a patient whose plasma contained relatively potent anti-Lua, active at 37°C, 51Cr-labelled Lu(a+) red cells (approximately 10 ml) survived normally (Greendyke and Chorpenning 1962); perhaps the antibody was solely IgM (non-complement binding) or IgG4. Anti-Lub has very rarely caused a DHTR (see Chapter 11) but is not known to have caused HDN (Lub is weakly expressed at birth). Examples of anti-Lub that were at least partly IgG have destroyed test doses of Lu(a+ b+) cells only slowly (Cutbush and Mollison 1958; Tilley et al. 1977), with more rapid destruction of Lu(a− b+) cells (Cutbush and Mollison 1958; Peters et al. 1978). An IgG1 anti-Lu6 destroyed 80% of a test dose of incompatible red cells in 1 h; results with 51Cr and flow cytometry agreed closely (Issitt et al. 1990).
An autoantibody to a high-frequency Lu antigen was associated with AIHA; the transfusion of 150 ml of least-incompatible blood caused chills, fever, dyspnoea and haemoglobinuria (Fitzsimmons and Caggiano 1981).
The Lutheran glycoprotein binds the basement membrane protein laminin 511/521. This interaction is between a binding site on Lutheran glycoprotein involving Asp312 on domain 3 and a surrounding cluster of negatively charged amino acids (Mankelow et al. 2007) and the alpha 5 chain of laminin (Kikkawa et al. 2007). Elevated levels of Lutheran glycoprotein are found on red cells from sickle cell patients (Udani et al. 1998) and, as sickle red cells adhere to alpha 5 chain containing laminin preparations (Hillery et al. 1996), it is possible that adherence of sickle red cells to laminin may be relevant to the vaso-occlusion occurring in vivo in patients with sickle cell disease.
Red cells from patients with polycythaemia vera have increased adhesive properties attributed to interaction between Lutheran glycoprotein on the red cells and laminin on endothelial cells (Wautier et al. 2007).
The Di, Yt, Xg, Sc, Do, Co, Ch/Rg, Ge, Cr, Kn, In, Ok, Raph, JMH and GIL Systems
Diego (Di)
The Diego system consists of two pairs of antithetical antigens, Dia and Dib, and Wra and Wrb, and several low-frequency antigens, Wda, Rba, WARR, ELO, Wu, Bpa, Moa, Hga, Vga, Swa, BOW, NFLD, Jna, KREP, Tra, Fra and SW1, all carried on band 3 and the result of single amino acid substitutions in the protein sequence (reviewed in Daniels 2002).
Dia and Dib
The antibody revealing the existence of the antigen Dia was found in a woman whose infant was affected with HDN (Layrisse et al. 1955). It soon became apparent that Dia is relatively common (frequency 5–15%) in South American Indians and in most Mongoloid populations but is very rare in white people. Surprisingly, it has been found in about 0.5% of Poles, possibly reflecting invasion by Tartars (Kusnierz-Alejska and Bochenek 1992). than 99.9% of white people are Di(b+); no Di(a− b−) subjects have been recognized through blood grouping studies. However, three cases of homozygous inactivating mutations in band 3 gene (SLC4A1) have been described all causing severe hereditary spherocytosis (reviewed in Bruce 2008). One individual homozygous for a band 3 mutation Val488Met (Band 3 Coimbra) had a 98% loss of band 3 protein and repeated blood transfusion was required during early childhood because of severe anaemia (Ribiero et al. 2000). Extensive analysis of the red cell membranes of this child revealed, in addition to band 3 deficiency, deficiency of protein 4.2, GPA, GPB, RhAG, Rh polypeptides, CD47 and LW glycoprotein (Bruce et al. 2003). Band 3 Coucouronnes is accompanied by a 65% loss of band 3. Individuals with band 3 Coimbra and band 3 Coucouronnes also have distal renal tubular acidosis (band 3 is expressed in erythroid cells and kidney).
The individual with band 3 Neapolis has an 88% loss of band 3 in red cells, but lacks distal renal tubular acidosis because the mutation does not affect band 3 expression in kidney. The severe disease phenotypes associated with homozygosity for these band 3 mutations probably explain why the Di(a− b−) phenotype has not been recognized through blood grouping studies, since in evolutionary terms homozygosity for inactivating mutations in band 3 would be incompatible with life. Hereditary stomatocytosis results from heterozygous inheritance of inactivating band 3 mutations. In this case the red cells have an increased permeability to cations (Bruce 2008).
The number of Dib sites on DibDib red cells was estimated to be 15 000 per cell (Masouredis et al. 1980b), although there are about 1 × 106 copies of band 3 per red cell. The substitution of Pro for Leu at amino acid 854 determines Dib rather than Dia (Bruce et al. 1994). Dib was not detected on lymphocytes, monocytes or granulocytes (Dunstan 1986).
Most examples of anti-Dia and anti-Dib are found in association with pregnancy; both antibodies are often IgG1 + IgG3 (Zupanska et al. 1990). A naturally occurring example of anti-Dia has been described (Steffey 1983) but anti-Dib seems always to be immune; the first two examples could be detected only by IAT and, apparently, their reactions were not enhanced by complement (Thompson et al. 1967).
Clinical Aspects
Anti-Dia is known to have been responsible for many cases of HDN but has not been definitely shown to have caused an HTR. Anti-Dib has only once been shown to have caused serious HDN (Ishimori et al. 1976), and infants may be completely unaffected (Habash et al. 1991). Only one HTR (delayed) has been described (Thompson et al. 1967).
Wra and Wrb
Wra and Wrb are antithetical antigens (Wren and Issitt 1988). Wra has an incidence of about 0.1% in white people; its incidence in other populations has not been extensively studied. All Wr(a−) subjects are Wr(b+), with the exception of those subjects who are GPA deficient. Wrb is a site of interaction between band 3 and GPA. A mutation at amino acid 658 (Glu → Lys) on band 3 defines Wrb and Wra respectively (Bruce et al. 1995). Mutations Gln63Lys (Jarolim et al. 1996) and Ala65Pro (Poole et al. 1999) in GPA cause abnormal Wrb expression. It is not known whether GPA is required for the expression of Wra. About 70 000 Wra sites were found on Wr(a+ b−) and Wr(a+ b+) red cells; the number of Wrb sites on Wr(a+ b+) cells was 150 000–350 000 and on Wr(a− b+) cells was 500 000 (Ring 1992).
Anti-Wra is a relatively common naturally occurring antibody, being found in about 1% of all blood samples. It is often found in subjects who have formed other blood group antibodies and in patients with AIHA. Anti-Wra often agglutinates red cells in saline, reacting more strongly at room temperature than at 37°C, but is frequently incomplete. Of 44 examples of anti-Wra
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


