Infectious agent
APS manifestations
Staphylococcus aureus
CAPS [142]
Streptococcus pyogenes
Escherichia coli
CAPS [143]
Klebsiella
CAPS [144]
Hepatitis C
Thrombosis, brain infarction [145]
Epstein-Barr virus
PE, thrombosis [146]
Parvovirus B19
Thrombosis [147]
Cytomegalovirus
HIV
Leg ulcer necrosis, arterial and venous thrombosis, vasculitis, livedo reticularis [149]
HSV
CAPS [150]
The Molecular Mimicry Hypothesis
Blank et al. [21, 22] hypothesized that molecular mimicry between infectious pathogens and β2GPI may serve as the origin of aPL and APS . Their theory was based on two lines of evidence: the striking association between APS and infectious agents and a strong amino acid similarity between β2GPI-derived peptides and various common pathogens (Table 3.2). Using a hexapeptide phage display library, they [21] identified several synthetic peptides, which exhibited high homology with proteins from the membrane particles of different bacteria and viruses, as target epitopes for monoclonal aβ2GPI derived from APS patients. For example, the sequence LKTPRV showed homology to eight different bacteria, such as Pseudomonas aeruginosa, and five kinds of viruses, such as human cytomegalovirus, while the sequence TLRVYK had homology to other bacteria and viruses. Furthermore, by neutralizing pathogenic aβ2GPI, these peptides inhibited both endothelial cell activation in vitro and induction of experimental APS in vivo.
Table 3.2
Candidate peptides with structural and functional similarity to the phospholipid-binding region of Domain V of β2GPI
Peptide | Source | Amino acid sequence | Inhibition of β2GPI binding to cardiolipin (%)a |
|---|---|---|---|
GDKV | Gly274-Cys288 in Domain V of human B2GPI | GDKVSFFCKNKKC | 43 |
GDKV2 | Modified GDKV with all six residues between Lys282 and Lys287 replaced with Lys | GDKVSFFCKKKKKKC | 56 |
TADL | Thr77-Glu96 of Adv type2 DNA-binding protein | TADLAIASKKKKKRPSPKPE | 68 |
TIFI | Thr101-Thr120 of ULB0-HCMVA from human CMV | TIFILFCCSKEKRKKKQAAT | 75 |
VITT | Val51-Ile70 of US27-HCMVA from human CMV | VITTILYYRRKKKSPSDT | 83 |
SGDF | Ser237-Ser256 of TLP-BACSU from Bacillus subtilis | SGDFEYTYKGKKKKMAFATS | N/A |
To demonstrate that molecular mimicry can trigger experimental APS, Blank and coworkers [7] immunized naïve mice with microbial pathogens that share structural homology with the TLRVYK hexapeptide. All immunized mice developed anticardiolipin antibodies (aCL) and aβ2GPI; the highest levels of aβ2GPI were found in mice immunized with Haemophilus influenzae, Neisseria gonorrhoeae, Candida albicans, or tetanus toxoid, while the lowest aβ2GPI levels were found in mice immunized with Klebsiella pneumoniae. Passive transfer of anti-TLRVYK antibodies (from immunized mice) into naïve mice resulted in thrombocytopenia, lupus anticoagulant (LA) activity, and increased fetal loss, i.e., experimental APS similar to mice injected with a pathogenic monoclonal aβ2GPI [7]. These findings demonstrate that bacteria with protein sequences homologous to β2GPI can induce aβ2GPI and APS manifestations [7], and provide evidence for a role for molecular mimicry in experimental APS.
Gharavi et al. [23] induced circulating aβ2GPI into naïve mice by immunizing with synthetic peptides derived from bacterial or viral proteins that show sequence similarity with a 15 amino acid peptide (GDKV) in the phospholipid-binding domain (Domain V) of β2GPI. The synthetic peptides included regions from the proteins of human adenovirus, cytomegalovirus, and Bacillus subtilis. Mice immunized with the peptides produced high levels of aCL and aβ2GPI, suggesting that viral and bacterial proteins may function like β2GPI and produce aPL through molecular mimicry of β2GPI. β2-glycoprotein I-derived synthetic peptides from regions other than Domain V, such as peptide NTLKTPRV from Domain I [21], also share sequence similarities with common bacterial antigens and interact specifically with aβ2GPI in mice, decreasing its thrombogenic potential [24]. Finally, the relationship between pathogens and β2GPI extends beyond sequence homology. β2-glycoprotein I binds to bacterial lipopolysaccharide (LPS), which is recognized by toll-like receptor 4 (TLR4) [25, 26], and functions as an in vivo scavenger of LPS [25]. The peptide sequence in Domain V responsible for LPS binding is conserved in all mammals [25]. An association between TLR4 gene polymorphisms and APS has been reported [27].
Leprosy and Syphilis
The association of infection with aPL and APS has been most thoroughly investigated in two infectious diseases, leprosy and syphilis. Studies in these infections highlight the need for astute differential diagnosis and careful characterization of the observed aPL in determining the pathogenic role of infection-induced aPL.
Leprosy (M. leprae)
Clinical presentation of leprosy
Leprosy, which has a wide range of clinical presentations, is caused by infection with the acid-fast bacillus M. leprae. The most basic classification separates leprosy into “paucibacillary” and “multibacillary” forms [28]. Multibacillary leprosy patients have acid-fast bacilli visible on bacilloscopic studies, high anti-M. leprae antibody titers, and more disseminated disease. Paucibacillary leprosy patients have no acid-fast bacilli visible on bacilloscopic studies and are treated with a shorter course of anti-M. leprae antibiotics. Leprosy can be subdivided into five forms based on clinical and histopathologic findings: tuberculoid (TT), borderline tuberculoid (BT), borderline (BB), borderline lepromatous (BL), and lepromatous (LL) [29]. Individuals with BT, BB, and BL leprosy are at risk for a Type 1 reaction, an inflammatory response thought to relate to M. leprae antigen release upon introduction of anti-leprosy therapy [30]. In contrast, BL and LL leprosy are associated with a Type 2 reaction, called erythema nodosum leprosum (ENL), thought to relate to immune complex formation in the setting of high antibody and high M. leprae antigenic load [31].
Antiphospholipid antibody prevalence in leprosy
Ribeiro et al. [32] found that 49% of 158 leprosy patients were positive for any aPL (46.2% for aβ2GPI and 15.8% for aCL), with IgM being the predominant isotype (88% and 84%, respectively). Compared with primary APS patients, leprosy patients had a higher prevalence of aβ2GPI (46.2% [73/158] in leprosy versus 23.7% [9/38] in APS) and lower prevalence of aCL (15.8% [25/158] in leprosy versus 89.5% [34/38] in APS). The frequencies of aβ2GPI and aCL were the same in leprosy patients who had completed or were still receiving anti-leprosy therapy; the frequencies were not increased in patients with leprosy immune reactions. Ribeiro et al. [33] followed aPL titers in 37 leprosy patients for a mean follow-up of 66.8 months. Thirty-two (86%) remained positive: 84% for aβ2GPI and 19% for aCL. Antiphospholipid antibody prevalence was also high (78%) in a study of 51 LL and BB leprosy patients from Argentina without clinical APS [34]. The rates of seropositivity for specific aPL were 57% (aβ2GPI), 61% (aCL), and 69% (LA), mostly IgM. The rate of aPL positivity did not differ during or following treatment [34]. Leprosy patients with aPL had higher plasma levels of soluble adhesion molecules such as P-selectin than did patients without aPL. The authors postulate that this finding relates to aPL-mediated activation of vascular endothelium [35].
Some studies have reported much lower rates of aPL: 3% aβ2GPI and 37% aCL positivity in a cohort of 35 multibacillary leprosy patients from Egypt [36]. Because these patients had leprosy for 15.2 ± 9.2 years, it is possible that their leprosy and any associated reactions (including aPL) would have resolved. No APS or thromboembolic phenomena were reported in these patients. As antibody levels and immune complex levels decrease as bacterial burden decreases, it is important to consider the stage of the disease when assessing aPL and APS. To evaluate patients prior to anti-leprosy therapy, Baeza et al. [37] studied 30 untreated multibacillary LL patients, of whom 23 (77%) were positive for IgG aCL and 23 (77%) were positive for IgM aCL. Additionally, 25 (83%) of LL sera bound to non-bilayer lipid arrangements containing mycolic acid. Levels of aCL IgG and IgM correlated with antibody reactivity to non-bilayer phospholipid (r = 0.77 and r = 0.69, respectively, p < 0.0001). The authors hypothesize that antibodies to non-bilayer phospholipid may disrupt cellular membranes, leading to the release of potentially immunogenic cellular components, such as aCL.
Antiphospholipid antibodies and the risk of thrombosis in leprosy
The association of aPL with thrombosis in leprosy is unclear. The fact that leprosy and systemic lupus erythematosus (SLE) have similar clinical and laboratory findings may make differential diagnosis difficult [38]. One study [39] found that 16% of 100 patients with multibacillary leprosy had four or more diagnostic criteria for SLE, yielding an 84% specificity for the diagnostic criteria. Overall, 20% of the 100 patients had one or more detectable aPL (aCL IgG, aCL IgM, LA, or Venereal Disease Research Laboratory [VDRL] test). However, none of the 20 patients with aPL had a history of vascular thrombosis or pregnancy loss. In a study that included seven multibacillary leprosy patients with a history of APS, only two had elevated aβ2GPI (2 IgM, 1 IgG) after the thrombotic event [40]. A case report described a BT leprosy patient who developed bilateral toe gangrene [41]. The patient was positive for aCL IgM at presentation and after 6 weeks of therapy; however, ultrasound was negative for arterial and venous thrombi.
Deep vein thrombosis (DVT) is an emerging risk in patients with multibacillary leprosy who receive thalidomide and corticosteroids for ENL [42–46]. The typical sequence of events preceding DVT is treatment with multi-antibiotic therapy for M. leprae, development of ENL, initiation of treatment with a corticosteroid, addition of thalidomide with corticosteroid taper, and development of DVT during the cross-taper of thalidomide and corticosteroid. In one such case, aβ2GPI and aCL IgG were slightly elevated when the patient developed DVT after 2 months of thalidomide treatment. The antibody levels returned to normal 8 weeks after the DVT [42].
Lucio’s phenomenon , characterized by recurrent ulcerative lesions affecting mainly the lower extremities, is a severe and potentially fatal immune reaction that occurs in patients with LL. In one case report from Ecuador of Lucio’s phenomenon with APS, aCL IgM was positive, and dermal vessels were occluded by thrombi [47]. Other reports found aCL positivity with vasculitis, not thrombosis [48, 49]. Some reports lack aPL testing but suggest evidence of thrombosis [50]. Levy et al. [51] found that aCL were β2GPI dependent in only two of 33 (6%) individuals with leprosy, both of whom had Lucio’s phenomenon. Forastiero et al. [52] compared the thrombogenic properties of IgM antibodies isolated from patients with leprosy or APS (and high levels of aCL, aβ2GPI, and LA) in a murine model of thrombosis. They found that IgM aPL from leprosy patients did not have thrombogenic and pro-inflammatory effects in vivo, when compared to aPL from APS patients, and present this as data supporting their hypothesis that thrombosis risk may relate to aPL type.
A study using mutant β2GPI showed that APS-derived aPL bound better to Domain V-deleted than to Domain I-deleted β2GPI, whereas leprosy-derived aPL bound to both mutant forms [53]. Furthermore, an anti-Domain I monoclonal antibody inhibited binding of APS-derived, but not leprosy-derived, aPL to β2GPI [53]. This difference in binding specificity may explain, at least in part, the different thrombogenic potentials of APS- versus leprosy-derived aPL.
Antiphospholipid antibodies and genetic polymorphisms in the β2GPI-encoding apolipoprotein H (APOH) gene
A number of groups have examined whether genetic polymorphisms in the APOH gene (chromosome 17) that encodes β2GPI differ in leprosy patients that develop aPL and thrombosis. Brochado et al. [40] characterized four single nucleotide polymorphisms (SNPs) in APOH in a cohort of 117 leprosy patients (seven had a history of APS, as defined by aPL positivity and confirmed thrombosis) and 113 non-leprosy controls in Brazil. They reported increased Leu/Leu and Val/Val at the Leu247Val SNP site (rs4581) in the leprosy group. Moreover, the multibacillary leprosy group with positive aβ2GPI IgM had an increased frequency of Val/Val homozygosity compared to controls [40]. The frequency of the mutant allele Ser316 was also higher in this group. In contrast, the two other SNPs examined, Cys306Gly and Trp316Ser, did not show significantly different allelic frequencies in leprosy patients compared with controls. Interestingly, the allele frequency at Leu247Val did not vary in a cohort study of Polish individuals with or without APS, and there was no association of the Leu247Val genotype with aβ2GPI levels [54].
Syphilis (Spirochete Treponema Pallidum)
Serological evaluation of syphilis
Syphilis is caused by infection with the spirochete Treponema pallidum. The serologic diagnosis of syphilis is reviewed by Morshed and Singh [55]. Non-treponemal tests for syphilis detect antibodies produced by the host in response to damaged cells. One of these antibodies is directed to cardiolipin, which is released by both damaged human cells and spirochetes. These tests have a high false-positive rate, given the nonspecific nature of these antibodies. For this reason, treponemal tests are also included in the diagnostic evaluation for syphilis.
Antiphospholipid antibody prevalence in syphilis
In a study on aPL and infectious diseases in South Africa, the rates of aCL and aβ2GPI positivity in syphilis were lower than in leprosy (8% versus 29% and 28% versus 89%, respectively) [56]. However, stage of disease, concurrent medications, and other information were not detailed. A study of aPL in Brazil included 74 syphilis patients who were positive in both VDRL and fluorescent treponemal antibody (FTA) tests, with most having completed penicillin treatment [57]. The rate of aPL positivity in these syphilis patients was 18% aCL IgG, 13% aCL IgM, and 10% aβ2GPI. No thrombotic events were noted. Guerin et al. [58] reported on aβ2GPI incidence among patients with a positive VDRL but negative confirmatory test for syphilis; these “false-positive” patients had detectable aβ2GPI IgM (17%), IgG (17%), and IgA (33%).
Antiphospholipid antibodies and the risk of thrombosis in syphilis
Affinity-purified aCL from five syphilis patients did not have LA activity, and only one inhibited prothrombin-to-thrombin conversion in vitro [59]; in contrast, all APS-derived aCL had both activities. In phospholipid-binding assays, syphilis-derived aCL recognized cardiolipin, but not phosphatidylserine, while all APS-derived aCL recognized both phospholipids. The dependence of phospholipid binding on β2GPI was not indicated in this study. The authors suggest that the difference in phospholipid recognition explains why hypercoagulability is not observed in patients with syphilis. Early studies demonstrated that APS-, but not syphilis-, derived aCL requires β2GPI for binding to phospholipid [60, 61], but recent literature suggests that the distinction between “autoimmune” and “infectious” aPL is less absolute than previously believed [62]. Infection could trigger induction of pathogenic aPL in genetically predisposed individuals, and the resulting aPL may be heterogeneous in their dependency on β2GPI.
Binding affinity and antibody isotype also play a role in aPL/β2GPI interactions. Metzger et al. [63] found that sera from syphilis patients bind to β2GPI under low salt conditions, but not higher (300 mM) salt conditions, in which APS sera still show strong binding, indicating that the binding affinity of aβ2GPI in patients with syphilis is lower than that in APS patients. Together, the differences in aPL derived from syphilis versus APS patients support the low incidence of aPL-associated thrombosis in syphilis patients.
The Microbiome
The chronic triggers that sustain aPL in APS patients are elusive. Emerging data suggest that microbiota, commensal organisms that colonize human hosts, likely contribute to APS pathogenesis [64]. “Commensalism” is a symbiotic relationship between two organisms of different species in which one derives some benefit, while the other is unaffected. The microbiota colonizes every niche of the human body, including the oral mucosa, gastrointestinal tract, sinobronchopulmonary (respiratory) tract, skin, and urogenital tract [65]. The barrier organ with the largest diversity of microbiota is the gut, which will be the focus of this section; other niches may also harbor triggers of aPL.
Gut commensals influence many aspects of innate and adaptive immunity [66]. Most notably, commensals induce key murine CD4 helper T cell subsets implicated in autoimmunity [67]. Segmented filamentous bacteria, species that colonize the murine small intestine by firmly attaching to the epithelium, are capable of inducing Th17 cells that specifically recognize segmented filamentous bacteria antigens [68]. Colonization with human colonic Clostridia species (Cluster IX and XIVa) leads to differentiation of regulatory T cells (Treg) in gnotobiotic mice (germ-free animals colonized with a defined set of microbes) [69]. Dysregulation in both Treg and Th17 cells is a well-established cellular mediator of autoimmunity. Follicular helper T cells (Tfh), a CD4 T cell subset that supports B cell antibody production, are key promotors of humoral immunity and autoantibody formation [70]. Murine Tfh are dependent on the gut microbiota [71], and B cell development occurs not only in the bone marrow but also in the gastrointestinal tract of mice [72].
The gut microbiome , extensively characterized in health and in several immune-mediated diseases [65, 73], is currently under investigation in APS patients [74]. While causal relationships between the microbiome and autoimmune diseases are difficult to establish in human studies, progress has been made in animal models. Several murine models of autoimmune disease are modulated by gut microbiota [67]. These microbiota-related effects are best demonstrated through germ-free rederivation of such models. The most dramatic examples are the loss of spondyloarthropathy and colitis when HLA-B27 transgenic animals are kept germ-free [75]; in contrast, non-obese diabetic mice (prone to type 1 diabetes) are increasingly susceptible to autoimmune destruction of the pancreatic islet cells with increasing hygiene [76]; germ-free animals develop diabetes with an almost 100% incidence [77]. Environmental factors other than cleanliness, for instance, specific dietary components, can also impact the gut microbiota and therefore immune function and autoimmunity (Fig. 3.1) [67, 78–80]. Dietary effects on microbiota observed in other autoimmune diseases [81] may have a similar impact on APS.
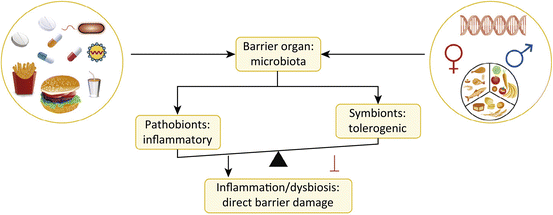
Fig. 3.1
Multiple factors influence the composition and function of the microbiota at barrier sites. The gut microbiota is profoundly affected by diet, medications, infectious agents, genetic makeup of the host, and also hormonal factors. These environmental and genetic influences shape the balance between pathobionts and symbionts, so that chronic inflammation ensues under certain autoimmune-prone conditions that leads not only to barrier damage (as shown in this figure) but also to distant autoimmune pathology, e.g., autoimmune thrombosis in various organs as in APS (The figure was adapted from Ref. [67])
Vieira et al. explored the gut microbiota in the (NZWxBXSB) F1 mouse, a spontaneous model of SLE and APS [81, 82]. Mice treated with an oral broad-spectrum antibiotic cocktail (vancomycin, metronidazole, neomycin, and ampicillin), or with vancomycin or ampicillin alone, did not develop serum aβ2GPI IgG or die from thromboemboli [82]. Bacterial load (16S ribosomal DNA) monitored in the feces of the mice was profoundly suppressed by broad-spectrum antibiotic treatment. These data suggest that gut commensals may play a role in the (NZWxBXSB) F1 model of APS.
Cell Death
Apoptotic cells provide a potential natural target and immunogen for aPL as well as for most autoantibodies found in SLE [83–86]. The apoptotic cell surface contains anionic phospholipid (phosphatidylserine) not present on the surface of viable cells [87–89], enabling the interaction of phospholipid-binding proteins such as β2GPI [90] and prothrombin [91], both important target antigens of aPL (Fig. 3.2). Interaction of β2GPI with apoptotic cells generates epitopes that are immunogenic in normal mice [92]. More recent findings indicate that β2GPI is highly immunogenic when presented in the context of innate immune activation, such as that induced by bacterial LPS [93]. In fact, mice immunized with β2GPI not only develop high levels of aβ2GPI and aCL but also multiple SLE-related autoantibodies and lupus-like glomerulonephritis [93]. Salem et al. [94] showed that epitope spread in the autoantibody response from β2GPI to multiple autoantigens is associated with a strong T cell response to β2GPI, independent of the particular β2GPI T cell epitope specificity. The authors propose that B cells specific for apoptotic cell-associated surface autoantigens take up apoptotic cells via antigen-specific surface IgG, leading to surface MHC class II-associated presentation of multiple apoptotic cell-derived peptides, including those from β2GPI. In this way, a β2GPI-specific T cell can provide help to a B cell that internalizes an apoptotic cell with surface-bound β2GPI [93, 94].
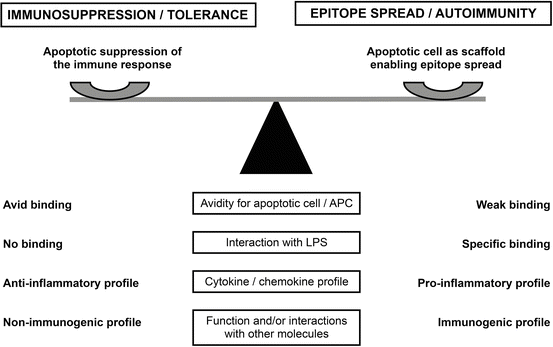
Fig. 3.2
Interaction of phospholipid-binding proteins with apoptotic cells: a balance between immunosuppression and autoimmunity. The hypothetical model proposes that the characteristics of a phospholipid-binding protein, and the nature of its interaction with apoptotic cells, can affect the balance between immunosuppression/tolerance and epitope spread/autoimmunity. On the one hand, apoptotic cells can suppress the adaptive response to an antigen when it is physically associated with the cells. On the other hand, apoptotic cells can serve as a scaffold that links surface-bound phospholipid-binding protein (e.g., β2GPI) to other molecules (particularly lupus-associated autoantigens) on the apoptotic cell. Multiple factors determine whether the balance is tipped toward immunosuppression/tolerance or toward epitope spread/autoimmunity. This figure shows several potential factors (affinity/avidity for apoptotic cell/APC, interaction with LPS, cytokine/chemokine profile, and function of the phospholipid-binding protein and/or its interactions with other molecules) (boxed text, center). Characteristics of the phospholipid-binding protein that tip the balance toward immunosuppressive are listed on the left side of the figure, while those promoting epitope spread and autoimmunity are listed on the right side. SLE, systemic lupus erythematosus; β 2 GPI, β2-glycoprotein I; LPS, lipopolysaccharide; APC, antigen-presenting cells (This figure is reproduced with permission from Levine et al. (copyright 2014. SAGE Publications) [151])
Recently, a form of cell death called NETosis has been implicated in several autoimmune diseases, including APS and SLE [95]. Neutrophil extracellular traps (NETs) are responsible for a form of cell death that is distinct from apoptosis or necrosis. Yalavarthi et al. [96] reported that freshly isolated neutrophils from patients with primary APS have higher levels of spontaneous NET release than do those from healthy control subjects. Furthermore, exposure of neutrophils from healthy controls to APS patient serum or IgG, or monoclonal aβ2GPI IgG, stimulates NETosis. Neutrophil extracellular traps may be immunogenic, as they present captured microorganisms and autoantigens in an inflammatory milieu that stimulates an immune response [97]. However, the role of NETosis in aPL production remains unclear.
The Immunological Response
Antigenic Factors
Relatively little has been uncovered regarding the ontogeny of pathogenic aPL, but it is clear that both genetic and environmental factors play a role in its production. The role of genetics in APS will be addressed in Chap. 4; HLA and non-HLA genes likely confer a baseline risk for aPL generation and APS, while various environmental factors may augment and intensify this risk [98].
Animal models can elucidate the nature of the inducing antigen(s) and other factors involved in aPL production. Although early experiments focused on immunization of animals with putative antigens, such as cardiolipin, phospholipid alone failed to induce high-titer pathogenic aPL [99]. The discovery that β2GPI, not phospholipid, was the main antigenic target of autoimmune aPL [100, 101] led to immunization with human β2GPI, combined with cardiolipin or adjuvant, and the successful induction of aPL [99, 102]. In some cases, these antibodies had pathogenic effects [99]. More recently, de Laat et al. [103] showed that murine or human β2GPI combined with cardiolipin, or misfolded β2GPI itself, can trigger antibody to β2GPI. Recombinant Domain I, but not Domains II–V, induced aβ2GPI. Together these findings suggest that β2GPI can be immunogenic when presented in a context in which immunogenic cryptic epitopes are exposed.
Another change in β2GPI that may expose cryptic epitopes is increased oxidative stress, which can occur with infection and other pathologies [104]. Ioannou et al. [105] showed that the proportion of protective free thiol β2GPI (which constitutes the majority of circulating β2GPI) is low in APS patients compared to other autoimmune patients and healthy controls. The higher proportion of oxidized β2GPI may result in limited ability to protect cells from oxidative stress [105] and increased T cell immunogenicity [106]. Thus oxidative stress might give rise not only to a more immunogenic β2GPI but also a procoagulant microenvironment.
Innate Immune Factors
While the antigenic stimulus in aPL induction is important, the immune environment in which antigenic exposure occurs may be equally or more critical. Infections potentially provide both necessary ingredients: antigen, and an innate immune stimulus. In addition to providing antigenic peptides that mimic β2GPI, infectious pathogens expose the host to TLR agonists like LPS, cytokine/chemokine release, and selective activation or destruction of lymphocytes [107–110]. Hence, an infectious organism may be a molecular mimic or, just as importantly, modulate the innate immune system.
Rauch and Levine [111, 112] suggest a hypothesis that highlights the central role of innate immune receptors, especially TLR4, in breaking tolerance. Mice immunized with β2GPI and LPS develop high levels of aβ2GPI, aCL, and multiple SLE-related autoantibodies, as well as glomerulonephritis [93, 94]. Pierangeli and coworkers [113] also showed that activation of TLR, in this case TLR7 and TLR9, induces aPL, as well as tissue factor production and thrombus formation, in autoimmune-prone PL/J mice treated with cytomegalovirus-derived peptides. All outcomes were highest in mice treated with both agonists. In SLE-prone MRLlpr/lpr mice, aPL titers were decreased in mice deficient in TLR7 alone, or both TLR7 and TLR9, but not TLR9 alone. These data support the hypothesis that innate immune receptors TLR4, TLR7, and possibly TLR9 are involved in the loss of tolerance to β2GPI.
Regulatory Immune Factors
The break-in tolerance among APS patients may involve Treg dysfunction [114]. Peripheral blood mononuclear cells from healthy donors treated with increasing concentrations of aPL showed changes in T cell subsets, compared to cells treated with control IgG [114]. T helper2 (Th2) and Th17 cell frequencies were increased, while Th1 and Treg cell frequencies were decreased. A subsequent study in primary APS patients reported a reduced frequency of CD4+CD25+foxp3+ Treg cells compared to controls [115]. These studies suggest that Th1/Th2 imbalance coupled with Th17 upregulation may play a role in aPL production and APS.
What Is Controversial and/or Unknown?
Natural Autoantibodies
Increasing evidence suggests that aPL, including aβ2GPI, belong to the natural antibody repertoire [116–119]. Natural antibodies are antibodies present in the circulation without prior infection, vaccination, other foreign antigen exposure, or passive immunization. Natural antibodies are often directed against highly conserved epitopes or structures present in many species, such as clusters of charged molecules. They typically bind with low affinity to ligands of varying chemical composition and can react with a number of unrelated antigens (including self-antigens). It is possible that natural antibody production is driven by bacteria living in the intestine [64]. Support for the identification of aPL as natural antibodies comes from the observation that normal healthy individuals without APS can have memory B cells that produce aPL [120].
As part of the innate immune system, natural antibodies can influence metabolic processes. In a series of elegant studies in mice, Fleming et al. showed that natural aβ2GPI are involved in complement-mediated mesenteric ischemia/reperfusion-induced injury [121, 122]. Additionally, natural aPL may be involved in acute graft rejection after renal transplantation [123]. Natural antibodies play a role in the clearance of apoptotic bodies; β2GPI and aβ2GPI may be essential for the clearance of these cell remnants [124]. Recent epidemiological studies in a large cohort of SLE patients have shown that aβ2GPI IgM protects against lupus nephritis [11]. In a recent study, de Mast and coworkers [12] extended these findings and showed that aβ2GPI IgM protects against stroke. Up to 5% of the healthy population has benign, low-affinity aPL in their circulation, a prevalence that increases with age [125]. These observations suggest that low-titer and low-affinity natural aβ2GPI present in healthy individuals may serve a protective role.
The natural antibody repertoire comprises two major subsets: an overt antibody population and a cryptic or latent population [126], the latter defined by unmasking in vitro with high salt solution, low pH, or oxidative agents. Thus sera negative for aPL can become positive after oxidation or heating to 56 °C [127, 128]. Apparently, slight modulations within the antibody binding site can change epitope recognition. As these studies have been done using serum or plasma, the role of serum/plasma factors cannot be excluded. The epitope recognized by pathogenic autoantibodies against β2GPI is also cryptic and is present in Domain I of β2GPI. Notably, this epitope has been completely conserved in mammalian evolution [25]. The factors promoting the transition of natural aβ2GPI from benign to pathogenic remain elusive.
Related posts:
 Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
 Mechanisms of Antiphospholipid Antibody-Mediated Pregnancy Morbidity
Mechanisms of Antiphospholipid Antibody-Mediated Pregnancy Morbidity
 Mechanisms of Antiphospholipid Antibody-Mediated Thrombosis
Mechanisms of Antiphospholipid Antibody-Mediated Thrombosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


