Fig. 24.1
Mechanism of action of oncolytic viruses: selective replication in cancer cells, tumor lysis, release of virus, and spread within cancer tissue (Reproduced from Kirn D et al. (2001) Nat Med 7: 781–787) [57]
On similar lines, the proposed resistance mechanisms to oncolytic virotherapy are the presence of neutralizing antibodies in the host and innate immune response against oncolytic viruses [16].
24.1.5 Types of Oncolytic Viruses
There are two types of oncolytic viruses: (1) viruses that preferentially replicate in cancer cells and leave nonpathogenic cells unaffected due to either dependence on oncogenic signaling pathways or heightened sensitivity to innate antiviral signaling and (2) viruses that have undergone genetic manipulation and/or been genetically engineered for insertion or deletion of genes necessary for replication in normal but not cancer cells. Autonomous parvovirus, myxoma virus (poxvirus), Newcastle disease virus (paramyxomavirus), reovirus, and Seneca Valley virus (picornavirus) are viruses that are specific to replication in cancer cells. On the other hand, viruses such as measles virus (paramyxomavirus), poliovirus (picornavirus), vaccinia virus (poxvirus), adenovirus, herpes simplex virus, and vesicular stomatitis virus are either genetically manipulated or engineered [14]. Several oncolytic viruses are currently being tested in clinical trials. The only oncolytic virotherapy to be USFDA approved is talimogene laherparepvec (T-VEC) derived from HSV-1 in the treatment of melanoma. Other therapies being developed are coxsackievirus (CVA21), adenovirus (K901, CG0070), vaccinia (JX-594, GM-CSF RV, Pexa-Vec), and reovirus (pelareorep, REOLYSIN®) [4, 15].
24.2 T-VEC
24.2.1 What Is T-VEC
24.2.2 Structure and Proposed Mechanism of Action of T-VEC
This oncolytic virus is derived from HSV-1. Given that HSV-1 is well characterized and its biology is well known, it serves as a suitable vector. Its large non-integrating double stranded DNA genome can be easily manipulated to insert large genetic inserts. The structure of T-VEC (Fig. 24.2) shows deletion of two HSV genes: ICP34.5 and ICP47. ICP34.5 confers neurovirulence to HSV-1. Thus, deletion of ICP34.5 provides selective cancer cell replication, and its replacement with two copies of the GM-CSF gene enhances local production of GM-CSF. GM-CSF is responsible for maturation and recruitment of dendritic cells and macrophages within the tumor, and these in turn allow for tumor antigen presentation leading to T-cell-mediated cytotoxic effect. Deletion of ICP47 causes an early activation of the US11 promoter which enhances viral replication and facilitates antigen presentation leading to better antitumor immune response [3, 15, 24].
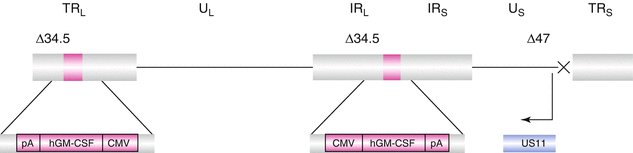
Fig. 24.2
Structure of T-VEC genome. Positions of deletions of ICP34.5 and ICP47 are marked as Δ34.5 and Δ47, respectively. The area marked in pink is the insertion site for the human GM-CSF gene (Reproduced from Hughes et al. (2014) Oncolytic Virotherapy 3: 11–20) [58]
Similar to other oncolytic viruses, T-VEC has been proposed to have both local and systemic effects (Fig. 24.3). Through mechanisms outlined above, T-VEC selectively replicates in tumor cells resulting in oncolysis. This in turn releases more progeny viruses and the cycle repeats itself. In conjunction with production of immunomodulatory cytokine GM-CSF, release of tumor-specific antigens induces recruitment and maturation of dendritic cells. Tumor cell antigen presentation to T cells occurs leading to activation and expansion of CD8+ T cells eliciting a systemic immune response [15].
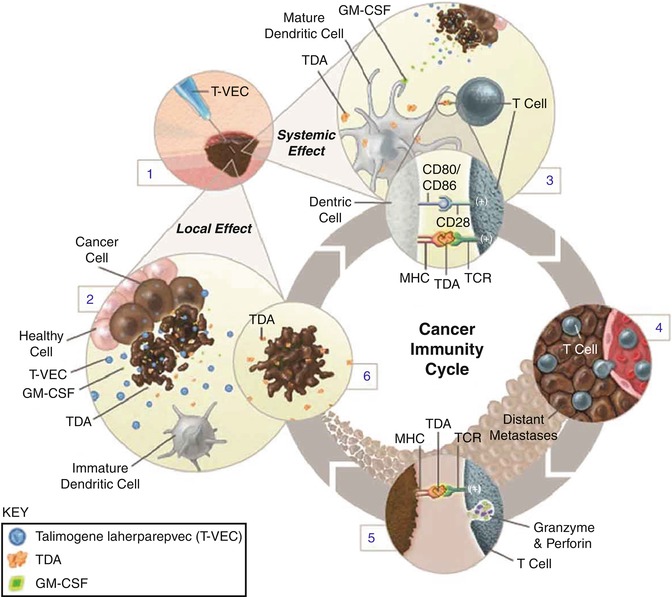
Fig. 24.3
Mechanism of action of T-VEC. After intratumoral administration of T-VEC (1), the virus replicates selectively in cancer cells (2). T-VEC also expresses GM-CSF, which promotes maturation and function of dendritic cells eliciting a systemic T-cell-mediated antitumor response (3). The virus can proliferate systemically to distant tumor sites (4). Tumor cell antigen presentation to T cells results in activation of innate immune system (5). Tumor cell lysis releases tumor-derived antigens (TDAs) (6) (Reproduced from Harrington et al. (2015) Expert Rev. Anticancer Ther 15: 1389–1403) [15]
24.2.3 Administration, Handling of T-VEC
A maximum injectable volume is determined based on the tumor dimension (Table). The volume of T-VEC administered varies by the lesion size: 0.1 ml for tumors up to 0.5 cm in longest dimension, 0.5 ml for tumors ranging from 0.5 to 1.5 cm, 1 ml for tumors ranging from 1.5 to 2.5 cm, 2 ml for tumors ranging from 2.5 to 5 cm, and 4 ml for tumors greater than 5 cm in longest dimension. The maximum volume per visit is 4 ml, and the volume to be administered is based on volume calculations for the tumor using its longest dimension. When multiple lesions are present, it is recommended that the largest lesions receive priority first followed by any lesions and then symptomatic lesions. Prior to injecting T-VEC, the area to be injected must be cleaned with alcohol swab and allowed to air-dry. If the lesion is not superficially palpable, then ultrasound guidance should be used for deeper subcutaneous tumors. Usually a single insertion site for the needle with T-VEC is recommended, but multiple sites may be necessary for larger tumors. Premedications and local anesthetics are usually not necessary prior to administration unless there is prior history of pain at the injection site. After treatment, holding pressure for 30 s at the site of injection is recommended. This should be covered with gauze and occlusive dressing and should remain covered for 1 week after each treatment. All materials in contact with the treatment site should be disposed of using universal precautions.
T-VEC is handled as a biosafety level 1 agent, which implies that it does not consistently harm normal healthy humans. It is prepared in a sterile biosafety cabinet. It is available in two different types of vials: the yellow-green vial with a concentration of 106 PFU/ml and the blue vial with a concentration of 108 PFU/ml. These are stored at −70°C or colder and thawed at room temperature until it converts to liquid form for use. Once thawed, the vials can be refrigerated for 12 h (106 PFU/ml concentration) to 48 h (108 PFU/ml concentration). Once thawed, the vials cannot be refrozen. Protective personal equipment with universal precautions must be followed while handling T-VEC. In case of accidental exposure, area should be cleaned with water for 15 min. In the event of a spill, 10% bleach solution should be used with absorbent materials to clean the surface [25].
24.2.4 Preclinical Studies Using T-VEC
A murine A20 tumor model was studied. HSV-1 viral strains with and without GM-CSF gene expression were injected into single side tumors in a bilateral flank system. HSV-1 viral strains both with and without GM-CSF expression resulted in tumor regression; however, only GM-CSF expressing HSV-1 resulted in tumor regression on the contralateral side. Cytotoxic T cells primed to parental A20 cells were seen only with treatment with HSV-1 with GM-CSF expression suggesting long-term antitumor immune response generated by this strain. This laid the foundation of further genetic modification of HSV-1 and the generation of T-VEC [3, 26].
Therapies such as T-VEC are considered ideal for melanoma treatment because (1) the presence of in-transit metastases denotes spread of melanoma cells to dermal lymphatics at the same time making surgical resection difficult and (2) metastases from melanoma preferentially spread to the skin making it optimal for targeting through intralesional therapies [24]. Therefore, much of the advancements in T-VEC therapy have been in melanoma. In keeping in line of intralesional therapies, agents such as BCG, GM-CSF, IL-2, PV-10, and IFN-alpha have been used and demonstrated responses in injected and noninjected lesions but in less than half of patients treated. Cutaneous toxicity and lack of systemic benefit have limited their use for treatment [24].
Administration of T-VEC was shown to increase local and systemic MART-1-specific CD8+ effector T cells, while at the same time there is decrease in both CD4+ Foxp3+ regulatory T cells and myeloid-derived suppressor cells (MDSC). Proportion of Tregs is lower in injected sites as compared to uninjected sites [3, 27].
24.2.5 Early Phase Clinical Trials
In a Phase I trial, T-VEC was evaluated for safety, optimal dose, and schedule in 30 patients including advanced breast cancer, head and neck cancer, colorectal cancer, and melanoma with cutaneous, subcutaneous, or superficial nodal sites of disease. Out of 19 evaluable histological specimens, inflammation and necrosis were noted in 73% of biopsies from the site of injection. The necrosis stained strongly for HSV protein. Tumor specificity of T-VEC was demonstrated by no evidence of necrosis in the non-tumor cells within the tumor microenvironment. The optimum dose and schedule derived from this study were initial dose of 106 PFU/ml followed by a dose of 108 PFU/ml after 3 weeks and repeated every 2 weeks until clinical response, toxicity, or disease progression. Clinical activity was similar between HSV-seronegative and HSV-seropositive patients [28].
After demonstration of good biologic activity of T-VEC in the Phase I study, a Phase II study was conducted in patients with stage III and IV melanoma. Twenty-six percent of patients achieved some form of response as assessed using RECIST criteria including eight patients achieving complete response. Durable responses were seen in 92% of patients ranging from 7 months to 31 months. The survival rate at 1 year was 58% in the intention to treat population, while it was 93% in those with initial objective response [3, 29].
In a Phase Ib study [1] (NCT01740297) conducted in the USA, 19 patients received T-VEC in combination with ipilimumab. T-VEC was administered at a dose of 106 PFU/ml on day 1 and second dose in 3 weeks at a dose of 108 PFU/ml repeated every 2 weeks. Ipilimumab was administered at a dose of 3 mg/kg every 3 weeks for four doses starting on day 1 of week 6. The regimen was overall well tolerated, except in five patients in which ≥Grade 3 adverse events (AE) were reported. Nausea was a common Grade 3 adverse event. Other AEs reported were fever, fatigue, flu-like symptoms, dehydration, diarrhea, and vomiting. Overall response rate assessed using the immune-related response criteria (irRC) [30] was 56%, with four (33%) complete responses and five partial responses. This overall response rate is comparable to 56% that was seen with ipilimumab and nivolumab, except that 55% of participants had Grade 3/4 AEs [31]. Durable response rate (responses seen for ≥6 months) was 44%. Twelve-month progression-free survival (PFS) was 50% and overall survival (OS) 72%. Activated CD8+ T cells (CD3+, CD4−, HLA-DR+) increased 1.5-fold from baseline after treatment and correlated with response.
On similar lines a Phase Ib/III study (NCT2263508) studying the combination of T-VEC with pembrolizumab in previously untreated, unresectable stage III/IV melanoma patients is being conducted. Based on results published so far from the Phase Ib portion of the study, the confirmed overall response rate as assessed using irRC was 57.1% with CR rate of 23.8%. Participants received T-VEC at a dose of 106 PFU/ml on day 1, 108 PFU/ml on day 22 and every 2 weeks thereafter, while pembrolizumab was administered at a dose of 200 mg IV on day 36 and every 2 weeks thereafter. Grade 3/4 AEs were noted in 33% of patients, and no Grade 5 AEs were noted [32].
In another Phase I trial studying the safety of T-VEC in advanced pancreatic cancer patients, it was demonstrated that T-VEC can be administered directly to visceral lesions via endoscopic, ultrasound-guided, fine needle injection (EUS-FNI) safely. The trial was designed such that patients would receive their first dose ranging between 104 PFU/ml and 106 PFU/ml followed by every 2 weeks of additional two doses of T-VEC ranging 107–108 PFU/ml all administered endoscopically. Four cohorts were planned; however, the study was stopped prior to enrollment of cohort 4 for reasons other than safety. Two of four in cohort 3 showed tumor reductions greater than 30%. Most common AEs observed were ascites, dehydration, anemia, abdominal pain, constipation, nausea, and vomiting. Although two Grade 5 AEs were noted, these were not attributed to the study drug. Interestingly, 59% of participants discontinued the study before receiving the planned treatment [33].
T-VEC has also been studied in combination with radiotherapy and cisplatin in a Phase I/II study in patients with untreated stage III/IV squamous cell cancer of the head and neck. Participants received chemoradiotherapy 70 Gy in 35 fractions with cisplatin dosed at 100 mg/m2 on days 1, 22, and 43. T-VEC was administered as an initial dose of 106 PFU/ml followed by two additional doses of 106 PFU/ml, 107 PFU/ml, or 108 PFU/ml in cohorts 1, 2, and 3, respectively. The regimen was overall well tolerated with no reported DLTs. All patients underwent neck dissection after 6–10 weeks. Response rate as assessed by RECIST was 82% (four complete responses, ten partial responses), and pathological complete response was noted in 93% of patients at the time of neck dissection. At 29 months, the disease-specific survival was 82% and relapse-free survival of 76% [34].
Other selected ongoing trials are listed in Table 24.1.
Table 24.1
Summary of selected ongoing trials with oncolytic virusesa
Trial name/ID | Phase, sponsor | Tumor type | Study population | Treatment | Primary end points | Secondary end points | Estimated number of participants |
|---|---|---|---|---|---|---|---|
Herpesvirus | |||||||
(A) T-VEC | |||||||
NCT02211131 | Phase II, Amgen | Melanoma | Resectable stage IIIB/C and IVA melanoma | Immediate surgical resection versus six doses of neoadjuvant T-VEC followed by surgical resection | Efficacy | PCR, OS, RFS, rate of R0 resection, overall response, safety | 150 |
NCT02453191 | Phase Ib/II, Amgen | Sarcoma | Locally advanced soft tissue sarcoma | T-VEC with concurrent preoperative external beam radiotherapy | Phase I—safety and tolerability Phase II—efficacy as assessed by PCR rates | ORR, TTP, OS | 32 |
NCT02509507 | Phase I, Amgen | HCC/liver metastases | Progressive HCC or liver metastases for solid tumors | T-VEC injected into liver tumors | Determine MTD | Safety | 100 |
MASTERKEY232/KEYNOTE-137/NCT026260000 | Phase Ib/III, Amgen | Squamous cell carcinoma of head and neck | Recurrent metastatic squamous cell carcinoma of head and neck | T-VEC in combination with pembrolizumab | Safety | Incidence of AEs, objective response rate, best overall response, duration of response, disease control rate, PFS, OS | 40 |
(B) HSV 1716 (SEPREHVIR) | |||||||
NCT00931931 | Phase I, Nationwide Children’s Hospital | Non-CNS solid tumors | Refractory localized or advanced non-CNS tumors from ages 7 to 30 | Intratumoral for localized disease or intravenous for metastatic disease of HSV 1716 | Safety | Measure antiviral immune response | 30 |
(C) HF10 | |||||||
NCT02272855 | Phase II, Takara BioVex Inc. | Melanoma | Unresectable or metastatic melanoma | Intratumoral HF10 plus intravenous ipilimumab | Best overall response rate | Safety, tolerability, objective response rate, PFS, durable response rate, 1-year OS, tumor-infiltrating lymphocytes, peripheral blood cytokines | 46 |
(D) G207 | |||||||
NCT02457845 | Phase I, UAB | Brain tumors | Children with progressive or recurrent supratentorial brain tumors | Catheter-infused G207 with or without single dose of radiation | Safety and tolerability | Immunologic response, viral shedding, PFS, OS, change in PS, QoL | 18 |
Reovirus | |||||||
(A) REOLYSIN® | |||||||
NCT02620423 | Phase I, Oncolytics Biotech | Pancreatic adenocarcinoma | Patients with advanced pancreatic adenocarcinoma who has progressed after (or did not tolerate) first-line treatment | REOLYSIN® and chemotherapy (gemcitabine or irinotecan or 5FU) in combination with pembrolizumab | Safety and DLTs | ORR, analysis of pre- and posttreatment biopsies and blood-based immune markers to determine effects of REOLYSIN® and pembrolizumab administered together | 9 |
NCT01274624 | Phase I, Oncolytics Biotech | Colorectal cancer | FOLFIRI naïve KRAS mutant metastatic colorectal cancer | Intravenous REOLYSIN® in combination with FOLFIRI and bevacizumab | DLTs, pharmacokinetic parameters for irinotecan and 5FU when combined with REOLYSIN® | CEA and objective response, clinical benefit rate, PFS, OS, safety, tolerability, correlative studies to identify biomarkers of response and efficacy | 32 |
Adenovirus | |||||||
(A) DNX-2401 | |||||||
CAPTIVE/KEYNOTE-192/NCT02798406 | Phase II, DNAtrix, Inc. | CNS tumors | Recurrent glioblastoma or gliosarcoma | Intratumoral DNA-2401 followed by pembrolizumab | Objective response rate | OS, time to response, duration of response | 48 |
(B) VCN-01 | |||||||
NCT02045602 | Phase I, VCN Biosciences, SL | Solid tumors | Advanced or metastatic solid tumors or pancreatic adenocarcinoma | Intravenous VCN-01 alone or in combination with nab-paclitaxel/gemcitabine | Safety, tolerability, RP2D | Viral genome analysis in tumor, viral pharmacokinetics, viral shedding, neutralizing antibodies anti-VCN-01, ORR, PFS | 36 |
(C) ONCOS-102 | |||||||
NCT02879669 | Phase I/II, Targovax Oy | Mesothelioma | Unresectable malignant pleural mesothelioma | Intravenous ONCOS-102 plus cyclophosphamide in combination with pemetrexed/cisplatin versus pemetrexed/cisplatin alone | Safety and tolerability | Determine and compare tumor-specific immunological activation. ORR, PFS, OS | 30 |
NCT03003676 | Phase I, Targovax Oy | Melanoma | Advanced or unresectable melanoma progressed after PD-1 blockade | Intravenous cyclophosphamide priming with intratumoral ONCOS-102 followed by pembrolizumab | Safety | Objective response rate, correlation of TILs and ORR, changes in immune subsets in tumor tissue and peripheral blood before and after ONCOS-102 and pembrolizumab, PFS, clinical benefit rate | 12 |
Vaccinia virus
Related posts: Peptide-Based Therapeutic Cancer Vaccines Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |||||||