In broad terms, viruses can cause cancer through either (or both) of two broad mechanisms: direct or indirect. Direct mechanisms, in which the virus-infected cell ultimately becomes malignant, are typically driven by the effects of viral oncogene expression or through direct genotoxic effects of viral gene products. In most established examples of direct viral oncogenesis, the cancerous cell remains “addicted” to viral oncogene expression for ongoing growth and viability.
A common feature of DNA viruses that depend on host cell DNA polymerases for replication (e.g., papillomaviruses, herpesviruses, and polyomaviruses) is the expression of viral gene products that promote progression into the cell cycle. A typical mechanism of direct oncogenic effects is through the inactivation of tumor suppressor proteins, such as the guardian of the genome, p53, and retinoblastoma protein (pRB). This effectively primes the cell to express the host machinery necessary for replicating the viral DNA. The study of tumor viruses has been instrumental in uncovering the existence and function of key tumor suppressor proteins, as well as key cellular proto-oncogenes, such as Src and Myc.
In theory, viruses could cause cancer via direct hit-and-run effects. In this model, viral gene products may serve to preserve cellular viability and promote cell growth in the face of otherwise proapoptotic genetic damage during the early phases of tumor development. In principle, the precancerous cell might eventually accumulate enough additional genetic hits to allow for cell growth and survival independent of viral oncogene expression. This would allow for stochastic loss of viral nucleic acids from the nascent tumor, perhaps giving a growth advantage due to the loss of “foreign” viral antigens that might otherwise serve as targets for immune-mediated clearance of the nascent tumor. Although hit-and-run effects have been observed in animal models of virally induced cancer,5 these effects are extremely difficult to address in humans. Currently, there are no clearly established examples of hit-and-run effects in human cancer.
In indirect oncogenic mechanisms, the cells that give rise to the malignant tumor have never been infected by the virus. Instead, the viral infection is thought to lead to cancer by attracting inflammatory immune responses that, in turn, lead to accelerated cycles of tissue damage and regeneration of noninfected cells. In some instances, virally infected cells may secrete paracrine signals that drive the proliferation of uninfected cells. At a theoretical level, it may be difficult to distinguish between indirect carcinogenesis and hit-and-run direct carcinogenesis, because, in both cases, the metastatic tumor may not contain any viral nucleic acids.
A variety of hunting approaches have been used to uncover etiologic roles for viruses in human cancer. The first clues that high-risk human papillomaviruses (HPVs), Epstein-Barr virus (EBV), Kaposi’s sarcoma–associated herpesvirus (KSHV), and Merkel cell polyomavirus (MCPyV) might be carcinogenic were based on the detection of virions, viral DNA, or viral RNA in the tumors these viruses cause. A common feature of known virally induced cancers is that they are more prevalent in immunosuppressed individuals, such as individuals suffering from HIV/AIDS or patients on immunosuppressive therapy after organ transplantation. This is thought to reflect the lack of immunologic control over the cancer-causing virus. Studies focused on AIDS-associated cancers provided the first evidence for the carcinogenic potential of KSHV and MCPyV. A theoretical limitation of this approach is that some virally induced cancers may not occur at dramatically elevated rates in all types of immunosuppressed subjects, particularly if the virus causes only a fraction of cases (e.g., HPV-induced head and neck cancers). Fortunately, the unbiased analysis of nucleic acid sequences found in tumors has become substantially more tractable as deep-sequencing methods have continued to fall in price. In the coming years, it should be increasingly possible to search for viral sequences without making the starting assumption that all virally induced tumors are associated with immunosuppression.6
One limitation of tumor sequencing approaches is that they might miss undiscovered divergent viral species within viral families known to have extensive sequence diversity7 and could miss viral families that have not yet been discovered.8 Tumor-sequencing approaches might also miss viruses that cause cancer by hit-and-run or indirect mechanisms. It is conceivable that this caveat could be addressed by focusing on sequencing early precancerous lesions thought to ultimately give rise to metastatic cancer.
An additional successful approach to hunting cancer viruses involves showing that individuals who are infected with a particular virus have an increased long-term risk of developing particular forms of cancer. This approach was successful for identifying and validating the carcinogenic roles of high-risk HPV types, hepatitis B virus (HBV), hepatitis C virus (HCV), KSHV, and human T-lymphotropic virus 1 (HTLV-1). Although viruses that are extremely prevalent, such as EBV and MCPyV, are not amenable to this approach per se, it may still be possible to draw connections between cancer risk and either unusually high serum antibody titers against viral antigens or unusually high viral load. Relatively high serologic titers reflect either comparatively poor control of the viral infection in at-risk individuals or expression of viral antigens in tumors or tumor precursor cells.9,10
The finding that a virus causes cancer is good news, in the sense that it can suggest possible paths to clinical intervention. These can include the development of vaccines or antiviral agents that prevent, attenuate, or eradicate the viral infection and thereby prevent cancer; the development of methods for early detection or diagnosis of cancer based on assays for viral nucleic acids or gene products; or the development of drugs or immunotherapeutics that treat cancer by targeting viral gene products. Unfortunately, establishing the carcinogenicity of a given viral species is an arduous process that must inevitably integrate multiple lines of evidence.11 The demonstration that the virus can transform cells in culture and/or cause cancer in animal models provides circumstantial evidence of the oncogenic potential of a virus. All known human cancer viruses meet this criterion. However, it is important to recognize that viruses can theoretically coevolve to be noncarcinogenic in their native host (e.g., humans) and cause cancer only in the dysregulated environment of a nonnative host animal. This caveat may apply to human adenoviruses.
Finding that viral DNA is clonally integrated in a primary tumor and its metastatic lesions helps address the caveat that the virus might merely be a hitchhiker that finds the tumor cell a conducive environment in which to replicate (as opposed to playing a causal carcinogenic role). This caveat is also addressed by the observation that, in most instances, viruses found in tumors have lost the ability to exit viral latency and are functionally unable to produce new progeny virions. An unfortunate consequence of this is that vaccines or antiviral agents that target virion proteins (e.g., vaccines against high-risk HPVs or HBV) or gene products expressed late in the viral life cycle (e.g., herpesvirus thymidine kinase, which is the target of drugs such as ganciclovir) are rarely effective for treating existing virally induced tumors.
Demonstrating that a vaccine or antiviral agent targeting the virus either prevents or treats human cancer is by far the strongest form of evidence that a given virus causes human cancer. This type of proof has fully validated the causal role of HBV in human liver cancer. Compelling clinical trial data also show that antiherpesvirus therapeutics can prevent KSHV- or EBV-associated lymphoproliferative disorders, and that vaccination against HPV can prevent the development of precancerous lesions on the uterine cervix.
PAPILLOMAVIRUSES
History
The idea that cancer of the uterine cervix might be linked to sexual behavior was first proposed in the mid 19th century by Dominico Rigoni-Stern, who observed that nuns rarely contracted cervical cancer, whereas prostitutes suffered from cervical cancer more often than the general populace.12 Another major milestone in cervical cancer research was Georgios Papanikolaou’s development of the so-called Pap smear for early cytologic diagnosis of precancerous cervical lesions.13 This form of screening, which allows for surgical intervention to remove precancerous lesions, has saved many millions of lives in developed countries, where public health campaigns have made testing widely available.
Although observations in the early 1980s suggested the possibility of a hit-and-run carcinogenic role for herpes simplex viruses in cervical cancer,14 this hypothesis was abandoned in light of studies led by Harald zur Hausen. Low-stringency hybridization approaches revealed the presence of two previously unknown papillomavirus types, HPV16 and HPV18, in various cervical cancer cell lines, including the famous HeLa cell line.15,16 There is now overwhelming evidence that a group of more than a dozen sexually transmitted HPV types, including HPV16 and HPV18, play a causal role in essentially all cases of cervical cancer. HPVs associated with a high risk of cancer also cause about half of all penile cancers, 88% of anal cancers, 43% of vulvar cancers, 70% of vaginal cancers,2 and an increasing fraction of head and neck cancers (see the following). In 2008, zur Hausen was awarded the Nobel Prize for his groundbreaking work establishing the link between HPVs and human cancer.
The viral family Papillomaviridae is named for the benign skin warts (papillomas) that some members of the family cause. In the early 1930s, Richard Edwin Shope and colleagues demonstrated viral transmission of papillomas in a rabbit model system.17 Using this system, Peyton Rous and others showed that cottontail rabbit papillomavirus-induced lesions can progress to malignant skin cancer.18,19 This was the first demonstration of a cancer-causing virus in mammals, building on Rous’ prior work demonstrating a virus capable of causing cancer in chickens (the Rous sarcoma retrovirus).
Tissue Tropism and Gene Functions
Although papillomaviruses can achieve infectious entry into a wide variety of cell types in vitro and in vivo, the late phase of the viral life cycle, during which the viral genome undergoes vegetative replication and the L1 and L2 capsid proteins are expressed, is strictly dependent on host cell factors found only in differentiating keratinocytes near the surface of the skin or mucosa. Interestingly, a majority of HPV-induced cancers appear to arise primarily at zones of transition between stratified squamous epithelia and the single-layer (columnar) epithelia of the endocervix, the inner surface of the anus, and tonsillar crypts. It is thought that the mixed phenotypic milieu in cells at squamocolumnar transition zones may cause dysregulation of the normal coupling of the HPV life cycle to keratinocyte differentiation.
There are nearly 200 known HPV types.20 In general, each papillomavirus type is a functionally distinct serotype, meaning that serum antibodies that neutralize one HPV type do not robustly neutralize other HPV types. Various HPV types preferentially infect different skin or mucosal surfaces. Different types tend to establish either transient infections that may be cleared over the course of months, or stable infections where virions are chronically shed from the infected skin surface for the lifetime of the host. HPV infections may or may not be associated with the formation of visible warts or other lesions. High-risk HPV types, with clearly established causal links to human cancer, are preferentially tropic for the anogenital mucosa and the oral mucosa, are usually transmitted by sexual contact, rarely cause visible warts, and usually establish only transient infections in a great majority of exposed individuals. The lifetime risk of sexual exposure to a high-risk HPV type has been estimated to be >70%. Individuals who fail to clear their infection with a high-risk HPV type and remain persistently infected are at much greater risk of developing cancer. Polymerase chain reaction (PCR)-based screening for the presence of high-risk HPV types thus serves as a useful adjunct to, or even a replacement for, the traditional Pap test.21
A consequence of the strict tissue-differentiation specificity of the papillomavirus life cycle is that HPVs do not replicate in standard monolayer cell cultures. Papillomaviruses also seem to be highly species restricted, and there are no known examples of an HPV type capable of infecting animals.22 Thus, the investigation of key details of papillomavirus biology has relied almost entirely on modern recombinant DNA and molecular biologic analyses.
Papillomavirus genomes are roughly 8 kb, double-stranded, closed-circular DNA molecules (essentially reminiscent of a plasmid). During the normal viral life cycle, the genome does not adopt a linear form, does not integrate into the host cell chromosome, and remains as an extrachromosomal episome or minichromosome. All the viral protein-coding sequences are arranged on one strand of the genome. The expression of various proteins is regulated by differential transcription and polyadenylation, as well as effects at the level of RNA splicing, export from the nucleus, and translation. In addition to the late half of the viral genome, which encodes the L1 and L2 capsid proteins, all papillomaviruses encode six key early region genes: E1, E2, E4, E5, E6, and E7.
The master transcriptional regulator E2 serves as a transcriptional repressor, and loss of E2 expression (typically through integration of the viral episome into the host cell DNA) results in the upregulation of early gene expression. The most extensively studied early region proteins are the E6 and E7 oncogenes of HPV16 and HPV18. The E6 protein of high-risk HPV types triggers the destruction of p53 by recruiting a host cell ubiquitin–protein ligase, E6AP.23–25 Another important oncogenic function of E6 is the activation of cellular telomerase.26 A wide variety of additional high-risk E6 activities that do not involve p53 have been identified.27
Most E7 proteins, including those of many low-risk HPV types, contain a conserved LXCXE motif that mediates interaction with pRB and the related “pocket” proteins p107 and p130.28 Interestingly, the LXCXE motif is present in a wide variety of other oncogenes, most notably the T antigens of polyomaviruses and the E1A oncogenes of adenoviruses. The interaction of E7 with pRB disrupts the formation of a complex between pRB and E2F transcription factors, thereby blocking the ability of pRB to trigger cell cycle arrest.29 The E7 proteins of high-risk HPVs can also contribute to chromosomal mis-segregation and aneuploidy, which may in turn contribute to malignant progression.30 Like E6, E7 interacts with a wide variety of additional cellular targets, the spectrum of which seems to vary with different HPV types.27
Some papillomavirus types express an E5 oncogene, which functions as an agonist for cell surface growth factor receptors such as platelet-derived growth factor beta (PDGF-β) and epidermal growth factor (EGF) receptor.31 Because E5 expression is uncommon in cervical tumors, it is uncertain whether the protein plays a key role in human cancer.
Human Papilloma Virus Vaccines
Two preventive vaccines against cancer-causing HPVs, trade named Gardasil (Merck) and Cervarix (GSK), are currently marketed worldwide for the prevention of cervical cancer. Both vaccines contain recombinant L1 capsid proteins based on HPV16 and HPV18 that are assembled in vitro into virus-like particles (VLPs). Together, HPV16 and HPV18 cause about 70% of all cases of cervical cancer worldwide. Gardasil also includes VLPs based on HPV types 6 and 11, which rarely cause cervical cancer but together cause about 90% of all genital warts. The VLPs contained in the vaccines are highly immunogenic in humans, eliciting high-titer serum antibody responses against L1 that are capable of neutralizing the infectivity of the cognate HPV types represented in the vaccine. It appears that the current HPV vaccines may confer lifelong immunity against new infection with the HPV types represented in the vaccine.32 The vaccines elicit lower titer cross-neutralizing responses against a subset of cancer-causing HPV types that are closely related to HPV16 and HPV18.33 Although these cross-neutralizing responses can at least partially protect vaccinees against a new infection with additional high-risk types, such as HPV31 and HPV45, it remains unclear how durable the lower level cross-protection will be.33
Because L1 is not expressed in latently infected keratinocyte stem cells residing on the epithelial basement membrane, current HPV vaccines are very unlikely to eradicate existing infections.34,35 Like keratinocyte stem cells, cervical cancers and precursor lesions rarely or never express L1. Thus, the existing L1-based vaccines seem unlikely to serve as therapeutic agents for treating cervical cancer.
Three types of next-generation HPV vaccines are currently in human clinical trials. Merck has recently announced that a newer version of Gardasil, which contains VLPs based on a total of nine different HPV types, remained highly effective against HPV16 and HPV18 and also prevented 97% of precancerous cervical lesions caused by a wider variety of high-risk HPV types.36 Another class of second-generation vaccines targets the papillomavirus minor capsid protein L2. An N-terminal portion of L2 appears to represent a highly conserved “Achilles’ heel”, which contains conserved protein motifs required for key steps of the infectious entry process.37 Anti-L2 antibodies can neutralize a broad range of different human and animal HPV types, and thus, L2 vaccines are hoped to offer protection against all HPVs that cause cervical cancer, all low-risk HPV types that cause abnormal Pap smear results, as well as the full range of HPV types that cause skin warts. Finally, a wide variety of vaccines that seek to elicit cell-mediated immune responses against the E6 and E7 oncoproteins are aimed at a therapeutic intervention for the treatment of cervical cancer.38
Oropharyngeal Cancer
It is well established that tobacco products and alcohol cause head and neck cancer. In the late 1990s, Maura Gillison and colleagues noted a surprising number of new cases of tonsillar cancer in nonsmokers.39 Many of the tumors found in nonsmokers were found to have wild-type p53 genes, raising the possibility that the tumor might be dependent on a p53-suppressing viral oncogene (as seen in cervical cancer). Gillison and colleagues went on to show that nearly half of all tonsillar cancers contain HPV DNA, most commonly HPV16. Interestingly, HPV-positive oropharyngeal cancers tend to be less lethal than tobacco-associated HPV-negative tumors. This finding has important implications for treatment of HPV-positive head and neck cancers.40
Although the incidence of tobacco-associated head and neck cancer has been declining in recent decades due to decreased tobacco use, recent studies suggest an ongoing increase in the incidence of HPV-associated cancers of the tonsils and the base of the tongue. By 2025, the number of new HPV-induced head and neck cancer cases in the United States is expected to roughly equal the number of new cervical cancer cases.39 Based in part on these observations, the U.S. Centers for Disease Control and Prevention recommends that boys, in addition to girls, should be vaccinated against high-risk HPVs.
Nonmelanoma Skin Cancer
Epidermodysplasia verruciformis (EV) is a rare immunodeficiency that is characterized by the appearance of numerous flat, wartlike lesions across wide areas of skin. The lesions typically contain genus betapapillomaviruses, such as HPV5 or HPV8. EV patients frequently develop squamous cell carcinomas (SCC) in sun-exposed skin areas (suggesting that ultraviolet [UV] light exposure is a cofactor). It is also well established that other immunosuppressed individuals, such as organ transplant recipients and HIV-infected individuals, are at increased risk of developing SCC.41,42 Although the E6 and E7 proteins of betapapillomaviruses appear to exert a different spectrum of effects than the E6 and E7 proteins of HPV types associated with cervical cancer,43–45 Betapapillomavirus oncogenes can transform cells in vitro.46 Although these circumstantial lines of evidence suggest that infectious agents, such as Betapapillomaviruses, might play a causal role in SCC, recent deep sequencing studies have observed few or no viral sequences in SCC tumors.47 Although the results argue against durable direct oncogenic effects of any known viral species in SCC, an animal model system using bovine papillomavirus type 4 strongly suggests that papillomaviruses can cause cancer by hit and run mechanisms.5 Thus, the question of whether hit-and-run or indirect oncogenic effects of HPVs may be at play in human SCC remains open.
POLYOMAVIRUSES
History
In the early 1950s, Ludwik Gross showed that a filterable infectious agent could cause salivary gland cancer in laboratory mice.48 Later work by Bernice Eddy and Sarah Stewart showed that the murine polyoma (Greek for “many tumors”) virus caused many different types of cancer in experimentally infected mice.49 The discovery that murine polyomavirus could be grown in cell culture helped rekindle research interest in tumor virology and interest in the question of whether viruses might cause human cancer.
Like papillomaviruses, polyomaviruses have a nonenveloped capsid assembled from 72 pentamers of a single major capsid protein (VP1). Both viral families also carry circular dsDNA genomes. These physical similarities initially led to the classification of both groups into a single family, Papovaviridae. When sequencing studies ultimately revealed that polyomaviruses have a unique genome organization (with early and late genes being arranged on opposing strands of the genome) and almost no sequence homology to papillomaviruses, the two groups of viruses were divided into separate families.
In the early 1960s, Bernice Eddy, Maurice Hilleman, and Benjamin Sweet reported the discovery of simian vacuolating virus 40 (SV40), a previously unknown polyomavirus that was found as a contaminant in vaccines against poliovirus.50,51 SV40 was derived from the rhesus monkey kidney cells used to amplify poliovirus virions in culture.52 SV40 rapidly became an important model polyomavirus, and studies of its major and minor tumor antigens (large T [LT]and small t [ST], respectively) have played an important role in understanding various aspects of carcinogenesis. Despite significant alarm about the possible risk SV40 might pose to exposed individuals, a comprehensive, decades long series of studies have failed to uncover compelling evidence that SV40 exposure is causally associated with human cancer.53
Two naturally human-tropic polyomaviruses, BK virus (BKV) and John Cunningham virus (JCV), were first reported in back-to-back publications in 1971.54,55 BKV and JCV are known to cause kidney disease and a lethal brain disease called progressive multifocal leukoencephalopathy, respectively, in immunosuppressed individuals. Although both viruses can cause cancer in experimentally exposed animals, it remains unclear whether either virus plays a causal role in human cancer. Although BKV LT expression can frequently be observed in the inflammatory precursor lesions that are thought to give rise to prostate cancer,56 there is no evidence for the persistence of BKV DNA in malignant prostate tumors.57 There have been case studies finding BKV T-antigen expression in bladder cancer,58 and some reports have indicated the presence of JCV DNA in colorectal tumors. The long history of conflicting evidence concerning possible roles for BKV or JCV in human cancer is reviewed elsewhere.59,60
Merkel Cell Polyomavirus
In 2008, Yuan Chang and Patrick Moore reported their lab’s discovery of the fifth known human polyomavirus species, which they named Merkel cell polyomavirus (MCV or MCPyV) based on its presence in Merkel cell carcinoma (MCC).61 The discovery used an RNA deep sequencing approach called digital transcriptome subtraction. Using classic Southern blotting, this report demonstrated the clonal integration of MCPyV in an MCC tumor and its distant metastases. Many other labs worldwide have independently confirmed the presence of MCPyV DNA in about 80% of MCC tumors.11
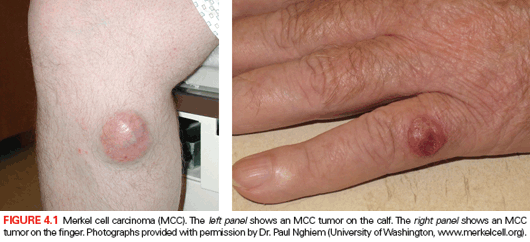
MCC is a rare but highly lethal form of cancer that typically presents as a fast-growing lesion on sun-exposed skin surfaces (Fig. 4.1).62 The risk of MCC is dramatically higher in HIV/AIDS patients, offering an initial clue that MCC might be a virally induced cancer.63 Although MCC tumors express neuroendocrine markers associated with sensory Merkel cells of the epidermis, one recent report has shown that some MCC tumors also express B-cell markers, including rearranged antibody loci.64 Currently, there is no clear evidence for the involvement of MCPyV in other tumors with neuroendocrine features.
In 2012, the International Agency for Research on Cancer (IARC) concluded that MCPyV is a class 2A carcinogen (probably carcinogenic to humans).10,53 It should be noted that IARC evaluations rely heavily on animal carcinogenicity studies, and the 2A designation was assigned prior to a recent report showing that MCV-positive MCC lines are tumorigenic in a mouse model system.65
A great majority of healthy adults have serum antibodies specific for the MCPyV major capsid protein VP1. A majority also shed MCPyV virions from apparently healthy skin surfaces, and there is a strong correlation between individual subjects’ serologic titer against VP1 and the amount of MCPyV DNA they shed.66–68 Interestingly, MCC patients tend to have exceptionally strong serologic titers against VP1.69 MCC tumors do not express detectable amounts of VP1, so this is unlikely to reflect direct exposure to the tumor and instead likely represents a history of a high MCPyV load in MCC patients. A recent study of archived serum samples shows that unusually high serologic titers against MCPyV VP1 often precede the development of MCC by many years.70
Like the LT protein of SV40 (and the E7 proteins of high-risk HPVs), an N-terminal portion of the MCPyV LT protein contains an LXCXE motif that mediates inactivation of pRB function. In contrast to SV40 LT, which carries a p53-inactivation domain that overlaps the C-terminal helicase domain, MCPyV LT does not appear to inactivate p53 function.71 Instead, the MCPyV LT helicase domain activates DNA damage responses and induces cell cycle arrest in cultured cell lines.72 This may explain why the LT genes found in MCC tumors essentially always carry mutations that truncate LT upstream of the helicase domain. siRNA experiments indicate that most (although possibly not all) MCC tumors are “addicted” to the expression of MCPyV T antigens.73–75 Interestingly, patients with higher levels of MCPyV DNA in their tumors, stronger T-antigen expression, and tumors that have been infiltrated by CD8+ T cells appear to have better prognoses.76 This is consistent with the idea that cell-mediated immunity can help clear MCC tumors that express MCPyV antigens.
Recent work has shown that the pRB interacting domain of LT mediates increased expression of the cellular gene survivin. The knockdown of survivin using siRNAs results in MCC tumor cell death and YM155, a small molecule inhibitor of survivin expression, protects mice from MCC tumors in a xenograft challenge system.77,78
In contrast to SV40, where LT appears to be the dominant oncogene, the MCPyV ST protein appears to play a key role in cell transformation. In addition to modifying the signaling functions of the cellular proto-oncogene PP2A, ST triggers the phosphorylation of eukaryotic translation initiation factor 4E binding protein 1.79 This results in dysregulation of cap-dependent translation and cellular transformation.
Although there is an intriguing epidemiologic correlation between MCC and chronic lymphocytic leukemia (CLL),80 there are conflicting reports concerning the presence of MCPyV in CLL and other lymphocytic cancers.81–83
Other Human Polyomaviruses
In recent years, the number of known human polyomaviruses has expanded dramatically. Of the 12 currently known HPyV species, only MCPyV has been clearly linked to human cancer. One new HPyV, trichodysplasia spinulosa polyomavirus (TSV or TSPyV) has been found in association with abnormal spiny growths on the facial skin of a small number of immunocompromised individuals.
EPSTEIN-BARR VIRUS
History
In 1958, Denis Burkitt provided the first clear clinical description of an unusual B-cell–derived tumor that frequently affects the jawbones of children in equatorial Africa.84 After hearing Burkitt give a 1961 lecture entitled “The Commonest Children’s Cancer in Tropical Africa – A Hitherto Unrecognized Syndrome,” Michael Epstein became interested in the idea that an insect vector-borne infection might account for the high incidence of Burkitt lymphoma in tropical Africa. Epstein, together with then PhD candidate Yvonne Barr, began examining tumor samples sent to them by Burkitt. Electron micrographs of lymphoid cells that grew out of the tumors in culture revealed viral particles with a morphology strikingly similar to herpes simplex viruses.85 It was soon shown that Epstein-Barr herpesvirus (EBV, later designated human herpesvirus 4 [HHV-4]) can transform cultured B cells and is the agent responsible for infectious mononucleosis.86–88
Although the initial conjecture that tropically endemic Burkitt lymphoma depends on a geographically restricted infectious agent ultimately proved correct, it was quickly established that the EBV infection is not restricted to the tropics. It instead appears likely that the malaria parasite Plasmodium falciparum is a key geographically restricted cocarcinogen responsible for endemic Burkitt lymphoma.53 In areas where children suffer repeated malaria infections, it appears that the parasite triggers abnormal B-cell responses, as well as weakened cell-mediated immune function, and these effects of recurring malaria infection in turn promote or allow the development of EBV-induced Burkitt tumors.11
Epstein-Barr Virus Life Cycle
EBV chronically infects nearly all humans. In a great majority of individuals, the infection is initially established in early childhood and is never associated with any noticeable symptoms. The infection is typically transmitted when virions, shed in the saliva of a chronically infected individual, come in contact with the oropharyngeal epithelium of a naïve individual. Although infected epithelial cells, such as keratinocytes, might serve to amplify the virus in some circumstances,89 the establishment of chronic infection is ultimately dependent on mature B cells, as subjects with X-linked agammaglobulinemia (who lack mature B cells) appear to be immune to stable EBV infection.90 Individuals who escape infection during childhood and instead first become infected during adolescence or adulthood often develop mononucleosis, which is associated with fevers and extreme fatigue lasting for weeks or sometimes months. Interestingly, late-infected individuals who experience mononucleosis and high EBV viral load are at increased risk of developing EBV-positive Hodgkin lymphoma.91
EBV-infected B cells can either go on to produce new virions, which are typically associated with cell lysis, or the virus can enter a nonproductive state known as latency. Viral latency is defined as a condition in which the virus expresses few (or possibly no) gene products but can, under some conditions, “reawaken” to express the full range of viral gene products and produce new progeny virions. Latently infected cells are highly resistant to immune clearance.
There are three recognized forms of EBV latency. In latency I, EBV nuclear antigen-1 (EBNA1), which is required for the stable maintenance of the circularized viral DNA minichromosome, is the only viral protein expressed. EBV-derived microRNAs (miRs) may also be expressed. At the other end of the spectrum, latency III is characterized by the expression of EBNA1–6, several latent membrane proteins (LMP1, 2A, and 2B), two noncoding RNAs (EBER1 and 2), the BCL-2 homolog BHRF1, BARF0, and multiple miRs. Although the initial discovery of EBV involved the visualization of virions, indicating that the virus had exited latency and entered the productive lytic phase of the life cycle, viral gene expression in EBV-induced cancers generally follows one of the three latent patterns. The oncogenic activities of various EBV gene products have recently been reviewed.87,88
In a great majority of healthy individuals, EBV exists almost exclusively in a latent state, with the occasional asymptomatic shedding of virions in the saliva. The infection is controlled, at least in part, by CD8+ T cells specific for various latency proteins. EBV, like other herpesviruses, expresses a variety of proteins that interfere with cell-mediated immune responses. Intriguingly, results from mouse model systems suggest that the chronic immunostimulatory effects of persistent gammaherpesvirus emergence (or abortive emergence) from latency in healthy hosts can nonspecifically boost immunity to other infections.92
Lymphomas
In addition to endemic Burkitt lymphoma, EBV is often present in sporadic cases of Burkitt lymphoma in individuals who have not been exposed to malaria. Although nearly all cases of endemic Burkitt’s lymphoma contain EBV DNA in the tumor (typically in a latency I–like state), only about 20% of sporadic cases arising in immunocompetent individuals contain EBV. Rates of Burkitt lymphoma are elevated in HIV-infected individuals, and HIV-associated Burkitt lymphomas contain EBV in about 30% of cases.
A common hallmark of all types of Burkitt’s lymphomas is deregulation of the cellular Myc proto-oncogene. A classic mutation involves chromosomal translocation of the Myc gene to the antibody heavy chain locus. Burkitt’s lymphoma tumors that lack detectable EBV DNA tend to carry multiple additional mutations in host cell genes, raising the possibility that an originally EBV-positive precursor cell ultimately accumulated mutations that rendered it independent of viral genes.88,93
In addition to Burkitt lymphoma, EBV is associated, to varying extents, with a histologically diverse range of other lymphoid cancers, including Hodgkin lymphoma, natural killer (NK)/T-cell lymphoma, primary central nervous system (CNS) lymphoma, and diffuse large B-cell lymphoma. The incidence of these various forms of lymphoma is significantly increased both in AIDS patients as well as in iatrogenically and congenitally immunosuppressed individuals.88 In particular, the essentially universal presence of EBV in CNS lymphomas in AIDS patients makes it possible to diagnose the disease with a PCR test for EBV that, together with radiologic findings, can obviate the need for a brain biopsy.
EBV is almost invariably associated with lymphoproliferative disorders, such as plasmacytic hyperplasia and polymorphic B cell hyperplasia, which are often observed in organ transplant recipients. These polyclonal lymphoproliferative responses can, in some instances, progress to oligoclonal or monoclonal lymphomas of various types. The occurrence of EBV-associated lymphoproliferative disease in immunosuppressed patients is generally heralded by the increased detection of EBV DNA in the peripheral blood and the oral cavity. This presumably reflects the failure of cellular immune responses to drive the virus into full latency and perhaps also a failure of cell-mediated immune responses targeting latency-associated EBV gene products present in the nascent tumor.
Carcinomas
In Southern China, NPC affects 25 out of 100,000 people, accounting for 18% of all cancers in China as a whole.94
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


