Fig. 1
Histological characteristics of CP subtypes HE stained section of ACP shows wet keratin (hash symbol), the palisaded cells (asterisk) circumscribing the tumour and the reticulo stellate cells (tilde symbol). These histological characteristics are magnified on the right side. Rosenthal fibres in the adjacent brain tissue are labelled by an arrow. PCPs consist of compact squamous epithelium
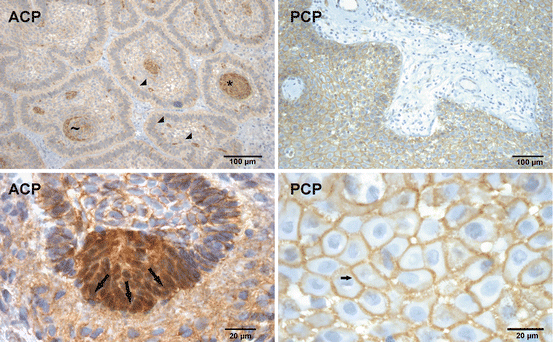
Fig. 2
Immunohistochemistry of β-catenin in CP . Nuclear β-catenin accumulation was found in most of the ACPs, in whorl like structures (asterisk) or in single cells (filled triangle). Note that not all cells are configured in whorls always show nuclear β-catenin enrichment (tilde). The lower figure illustrates the detection of β-catenin in whorl like structures forming a tip of outgrowing tumour cells (arrows). The nuclear accumulation is absent and β-catenin occurrence is restricted to the cell membrane (up arrow) in PCP
ACPs exhibit striking morphological and histological similarities to odontogenic tumours of the jaw. Most analogies were found to calcifying odontogenic cysts featuring proliferating ameloblastic epithelium with calcifications, wet keratin, ghost cells and cyst formations [16–19]. ACPs can also resemble ameloblastoma or adenomatoid odontogenic tumours [17, 20]. Moreover, odontogenic epithelial differentiations of ACPs are demonstrated by the detection of enamel proteins as amelogenin, enamelysin and enamelin which are exclusively expressed in teeth. This observation underscores the assumption that the ACP variant arises from embryonic remnants of the stomodeum representing Rathke’s pouch and teeth forming progenitor tissue [21, 22]. This is substantiated by the finding of a rudimentary tooth in an ACP of a 4-year-old boy [20]. It is worth noting that no similarity to odontogenic tumours could be established in PCPs [16, 17].
Genetic Distinctions of CP Subtypes
Genetic aberrations in PCP were first identified in 2014 by Brastianos et al. using exome sequencing [23]. The BRAF V600E mutation could be found in up to 100% of PCP analyses [7, 23, 24].
Sekine et al. first described activating mutations of the β-catenin encoding gene CTNNB1 in ACPs as early as 2002 [6]. However, until now, there was no CTNNB1 mutation described in PCP [6, 7, 23, 25].
Nearly all studies identified a distinctive occurrence of CTNNB1 mutations in ACP and BRAF mutations in PCP. However, one study showed the coexistence of both mutations in ACP [26]. It is worth noting that negative mutational screening is also sufficient to differentiate Rathke’s Cleft cyst with epithelial metaplasia from PCP which sometimes bear a histologically striking resemblance [24, 27, 28].
In ACP it was assumed that CTNNB1 mutations lead to nuclear accumulation of β-catenin all over the tumour. Interestingly, only distinctive cells were identified in ACPs revealing β-catenin accumulations [6]. This was observed to occur in up to 94% of this CP subtype [25]. Neoplastic cells with nuclear accumulation of β-catenin seemed not to be equally distributed throughout the tumour (Fig. 2). They were prevalently identified either in the centre of epithelial whorls or adjacent to ghost cells. However, whorl like configured cells do not always consistently express nuclear β-catenin (Fig. 2). There are also single cells, dispersed throughout the tumour exhibiting nuclear β-catenin. In contrast, nuclear accumulations within palisaded cells are rare and sometimes seen in the outer epithelial margin bordering either the brain parenchyma or mesenchymal scarring (Fig. 2).
Further lesions of the sellar region such as pituitary adenomas, PCPs, arachnoidal cysts, Rathke’s cleft cysts and xanthogranulomas do not show such distinct β-catenin accumulation as illustrated for ACP (Figs. 2, 5 and 7). This finding makes nuclear β-catenin a diagnostic hallmark of ACPs, setting them apart from PCP and other lesions that originate in the sellar region [25, 29].
β-catenin: A Multifunctional Protein and Key Component of the Canonical Wnt Signalling Pathway
The CTNNB1 gene encodes the 88 kDa protein β-catenin, which plays a crucial role in intercellular adhesion and is a central component of the canonical Wnt signalling pathway . Usually β-catenin is located at the cell membrane where it interacts with cadherins establishing adherence junctions (Fig. 3) [30]. Epithelial cells express E-cadherin, the prototype and best analysed cadherin of this transmembranous glycoprotein superfamily. Attachment to neighbouring cells is maintained by the extracellular domain of E-cadherin in a calcium dependent homophilic manner. It is anchored intracellularly to the cytoskeleton via β−catenin and α-catenin, which link the whole complex to actin (Fig. 3). Maintenance of cellular adhesion is important to keep cellular polarity, differentiation and tissue integrity [31]. However, distribution of β-catenin within the cell is adjustable and can vary rapidly from membranous anchorage to cytoplasmic occurrence impairing adherence junctions [32]. Dissociation of β-catenin from the adherence complex could be provoked by the epidermal growth factor receptor (EGFR) or src driven phosphorylation at Tyrosine of codon 654 (Y654) which reduces its binding affinity to E-cadherin [33, 34]. Fyn, Fer or c-Met promote phosphorylation of β-catenin tyrosine residue 142, which results in dissociation of α-catenin from β-catenin (Figs. 3 and 4) with concomitant loss of cadherin adhesion [33]. In the cytoplasm, β-catenin is tagged for proteosomal degradation, when Wnt signalling is inactive (Fig. 3) [35]. Its cytoplasmatic amount is thus strictly regulated by a multiprotein-complex containing the tumour suppressor adenomatous polyposis coli (APC), the scaffold proteins Axin and Axin2, the phosphokinases GSK-3β and casein kinase 1 (CK1). Following binding to this destruction-complex, β-catenin becomes phosphorylated within its N-terminal region encoded by exon 3 of the β-catenin gene (CTNNB1) (Figs. 3 and 4) [36–38]. The phosphorylation of Threonine and Serine residues in this region occurs in a well-regulated manner. Initially CK1 phosphorylates β-catenin at a Serine (S) encoded by codon 45 (S45) which provides the GSK-3β priming site [39]. Following GSK-3β binding Threonine 41 (T41) and S37, S33 and S29 are subsequently modified by phosphorylation (Fig. 4). The phosphorylation of S45 and T41 can also be maintained by I kappa B kinase alpha (IKK-α) [40] independent of CK1 priming (Fig. 4). Phosphorylation of S33 and S37 residues is essential for the recognition of β-catenin by the β-TrCP complex (Fig. 3) [41, 42]. Multiple ubiquitin molecules are added covalently to Lysine residues of β-catenin, which is recognised by the proteasome leading to its subsequent degradation [41].
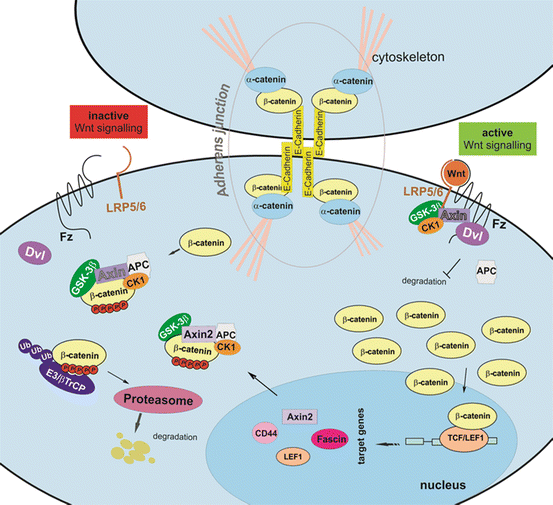
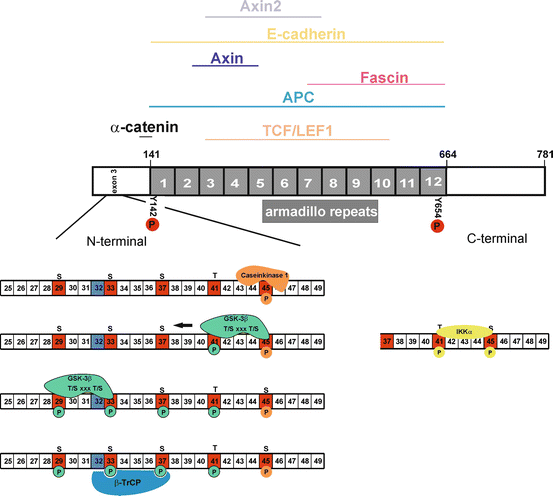
Fig. 3
Cellular β-catenin distribution and its regulation by Wnt signalling . The β-catenin protein is located at the cell membrane integrated in adherens junction complexes. The cytoplasmic pool is controlled by the destruction complex composed of APC, the scaffold proteins Axin/Axin2, as well as protein kinases CK1 and GSK-3β. The complex binds β-catenin and marks it by phosphorylation (red circles) for βTrCP driven ubiquitination leading to its subsequent proteosomal degradation. This process is inhibited if Wnt signalling is active. Wnt molecules bind to the frizzled (Fz) receptor and co-receptor LRP5/6. Dishevelled (Dvl) inhibits GSK-3β and therefore the designation of β-catenin proteosomal degradation. The protein accumulates within the cytoplasm and translocates to the nucleus. Here it possesses the function of a transcription co-factor by interacting with transcription factors of the TCF family [T-cell factor (TCF)/lymphocyte enhancer factor 1 (LEF1)] initiating target gene expression, e.g. Axin2, Fascin, CD44 and EGFR. Axin2 acts as a negative feedback regulator because it terminates Wnt signalling via acceleration of the β-catenin degradation
Fig. 4
β-catenin structure , its interaction sites with other proteins and its modifications. The central region of β-catenin consists of a 12 times repeated segment of 42 amino acids. This central part, also called armadillo repeats, is highly conserved and forms a positively charged groove which binds negatively charged proteins. A subset of proteins and their β-catenin interaction sites, which are depicted above, marked by a line. The functionality of β-catenin depends on its capacity to interact with different proteins, which is influenced by posttranslational modifications as phosphorylation (P). The N-terminal region in particular the proportion encoded by exon 3 is essential for β-catenin endurance. Protein kinases (casein kinase 1, IKKα and GSK-3β) phosphorylate Serine (S) and Threonine (T) residues establishing a β-TrCP binding site crucial for following ubiquitinylation targeting β-catenin for proteosomal degradation. Phosphorylation of Tyrosine (Y) codon 142 (Y142) and Y654 downregulates β-catenin interaction with α-catenin and E-cadherin, respectively. Modified from Xu, W. et al. (2007)
The activation of Wnt signalling (Fig. 3) leads via Dishevelled (Dvl) to membranous translocation of GSK3β , and the scaffold protein Axin [43], which in turn results in the decreased targeting of β-catenin for proteasomal degradation, accompanying with its cytoplasmic stabilisation and subsequent nuclear translocation [44]. Within the nucleus, β-catenin initiates gene expression via interaction with transcription factors of the LEF1/TCF family [31]. The target genes of β-catenin are promoters of cell proliferation, differentiation and also cell migration [32, 45].
Mutations in exon 3 (CTNNB1) are not Sufficient to Induce Nuclear β-Catenin Accumulation in ACP Per se
Molecular genetic studies of various ACP demonstrated a broad range of missense mutations within CTNNB1 exon 3, compromising residues essential for proteasomal degradation labelling, e.g. codons 32, 33, 34, 35, 37, 41, 43 and 45 (Fig. 4) [6, 7, 25, 26, 46]. In addition to point mutations, deletions in exon 3 were also observed [7, 23, 25].
The underlying cause leading to β-catenin accumulation only in distinct cells (Fig. 2) was evaluated in more detail. Activating mutations within exon 3 of the β-catenin gene (CTNNB1) were assumed to cause an increase of β-catenin levels within the cytoplasm and nucleus as a consequence of impaired degradation [6, 25]. If mutations within CTNNB1 promote cellular stabilisation of β-catenin, then it suggests that only those cells harbouring elevated β-catenin levels that also exhibit a particular genetic profile in relation to surrounding cells without β-catenin accumulations. Unexpectedly, as selective mutational analyses of β-catenin accumulating vs. non-accumulating cells revealed, both cell populations harbour activating mutations in exon 3 of the CTNNB1, without identifying a cell-specific mutation pattern [47]. These results imply that alterations in β-catenin gene regions, encoding phosphorylation sites and binding sites of β-TrCP-complex, which are crucial for protein degradation, are not sufficient to cause nuclear accumulation per se. In contrastto Sekine and colleagues, Kato et al. were not able to verify mutations beyond the epithelial tumour cell component [6, 46]. The identification of multiple mutations in a single CP sample [23, 47] suggests an increased genetic instability. However, chromosomal instability appears unlikely in CP [48]. As described in the following, the impact of β-catenin mutations on cellular transformation is associated with its degree of impaired degradation, depending on the affected codon of exon3. The majority of ACP mutations occur in codons 33 and 37, which were mostly found in benign entities, compared to codons 41 and 45 which are associated with more malignant transformed phenotypes [40]. Mutations in codons 32 and 34 do not display changes in GSK-3β phosphorylation, but decrease ubiquitination as they represent the ubiquitination recognition motive (Fig. 4). Cells with affected codon D32 were shown to be transformed to a higher degree in relation to S33 or G34 mutations, whereas resulting β-catenin accumulation does not increase target gene expression [42, 49]. It was shown that target gene expression occurs when β-catenin is not phosphorylated on Threonine 41 and Serine 37 [50]. Missense mutations of CTNNB1 codon 35 are seldom, and were only detected in hepatocellular carcinomas [51, 52]. This implies that identified mutations within the β-catenin gene exhibit various impacts on ACP tumourigenesis. Since these results revealed that β−catenin accumulations are not caused by mutations per se in exon 3 of the CTNNB1 gene, additional mechanisms have to be involved to achieve its cytoplasmic and nuclear stabilisation.
Many other mechanisms are likely to play a role, e.g. upregulation of β-catenin binding partners competing with multi-destruction complex component binding sites, thus preventing degradation (Fig. 4). Identified binding partners include TCF-family transcription factors, Axin2 and APC [53, 54]. In addition, armadillo repeat units 10–12 are essential for the passive nuclear-pore-complex translocation of β-catenin [55]. Although compromised nuclear shuttling of β-catenin may lead to nuclear accumulation, but no mutations within the CTNNB1 region encoding for the armadillo repeats (exon 8–13) were identified in ACP [47], thus indicating an intact passive nuclear transport machinery. Furthermore, a genetically impaired membranous anchorage of β-catenin facilitated by exon 4 [33] was also excluded [47]. This notion indicates that interaction of β-catenin with other proteins is not compromised by genetic alterations.
A large body of β-catenin binding partners , such as E-cadherin, LEF1, Axin/Axin2, APC and Fascin, [53, 54, 56–58] were identified as being expressed in ACPs. Importantly, proteins that share the same β-catenin binding sites (Fig. 4) compete with each other [59]. If binding of the destruction complex is permitted, β-catenin is stabilised [56]. In addition, binding partners regulate the subcellular localisation of β-catenin [60]. This finding supports the cellular reallocation of β-catenin in ACPs. Stabilisation of β-catenin, as a result of activated SHH or EGFR signalling, could provide an additional mechanism [61–64]. In summary, activating alterations in exon 3 of CTNNB1 are not restricted to nuclear β-catenin accumulating tumour cells.
It is important to note that the impact of compromised β-catenin in the genesis of ACP could be proven in a genetically modified mouse model [65].
Systematic analyses of the β-catenin associated Wnt signal transduction pathway offer the opportunity to unravel new insights into the pathogenesis of ACP. The current knowledge about the impact of β-catenin accumulating cells on cellular proliferation, differentiation and migration in ACP cells will be illuminated in the following.
ACPs Show Activation of Wnt Signalling Target Genes in Cell Clusters with Nuclear β-Catenin Accumulation
Cellular localisation of β-catenin is not per se sufficient to promote target gene activation [66]. However, it has been shown that nuclear β-catenin accumulations, as a hallmark of ACP, are sufficient to induce Wnt signalling target gene expression, e.g. Axin2, BMP4 and CD44 (Fig. 5) [7, 47, 67]. This was identified on mRNA as well as protein level in tumour cell clusters exhibiting nuclear β-catenin accumulations. Comparative gene expression analyses of both CP variants revealed that Axin2 and BMP4 are increased in ACP. Furthermore, the target gene Fascin1 [68] and EGFR (unpublished data) was identified as being regulated by β-catenin, using knockdown experiments in primary ACP cell cultures.
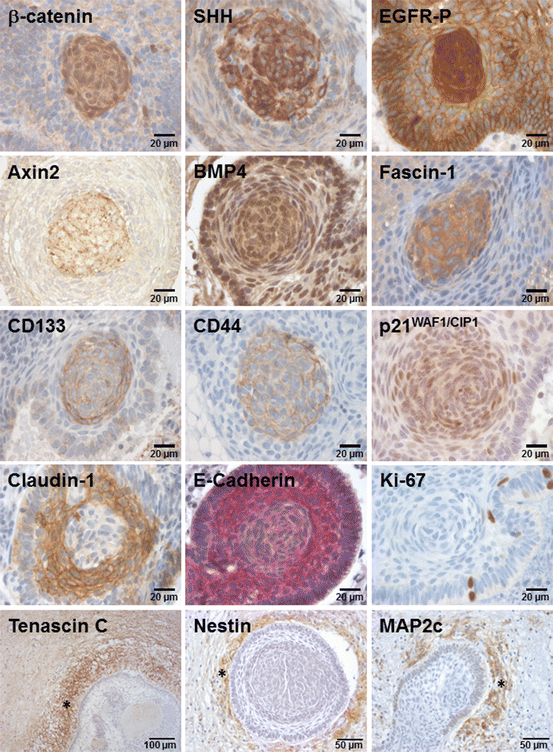
Fig. 5
Expression of proteins in relation to β-catenin accumulating whorls. Cells with nuclear β-catenin accumulation are often configured in a whorl shape. The cells with an activated Wnt signalling pathway also exhibit activated SHH and EGFR signal transduction. It was shown in various studies that the target genes (Axin2, BMP4 and Fascin-1), stem cell markers (CD133 and CD44) and the cell cycle inhibitor p21Waf1/CIP1 co-localise in cells with nuclear β-catenin. Proliferating Ki-67 positive cells were mostly identified in the palisading cell layer. Cell adhesion component expression, e.g. claudin-1 (Tight junctions) and E-cadherin (adherens junctions) were decreased in whorl like structures. The brain-tumour boundary (asterisk) is characterised by increased tenascin-C, Nestin and MAP2c levels. The letter could also be found in the picket fence-like cells
Axin2
Axin2 also known as conductin (mice) and Axil (rats) is a homolog of Axin. Axin2 is only detected in cells with activated Wnt signalling in contrast to Axin, which is expressed ubiquitously during development. Further studies revealed that Axin2 is a direct target of the Wnt pathway [69]. Its cellular function is comparable with that of Axin because it is also able to bind APC, GSK3β and β-catenin [36] (Fig. 3). Axin2 facilitates as a component of the destruction complex GSK 3β−driven phosphorylation and degradation of β-catenin and acts, therefore, as a negative feedback regulator of the Wnt signalling pathway. The expression of Axin2 is therefore an important mechanism in terminating the action of β-catenin [69, 70]. Interruption of Axin2 by mutations was shown to be associated with an enhanced Wnt signalling that causes tooth agenesis and tumour genesis [71]. The latter has been recognised, for example, in colon cancer, medulloblastomas and hepatoblastomas [72].
BMP4
The bone morphogenetic protein 4 (BMP4) is a secreted factor belonging to the transforming growth factor beta (TGF-β) superfamily. Mature BMP4s form dimers which bind to transmembranous serine/threonine kinase of the type I and type II TGF-β receptors, thereby composing multimeric receptors. The formation of these heteromeric complexes initiates a cascade of several Smad protein modifications. Finally, Smads translocate into the nucleus and co-activate gene expression [73]. The expression of BMP4 plays a crucial role during organogenesis in both development and cellular transdifferentiation. BMP4 knockout leads to early lethality as observed in mice embryos. It plays an important role as a signalling molecule in the development of the central and peripheral nervous system, musculature and skeleton. BMP4 is essential in the initiation of the pituitary gland [73–76]. The presumptive ventral diencephalon, which contacts the oral ectoderm (stomodeum), produces BMP4 and induces the formation of Rathke’s pouch primordium [75]. An interaction mediated by several cytokines between epithelial cells and surrounding mesenchyme leads to the generation of specialised tissues. BMP4 was also observed to play an important role downstream of Wnt signalling during early tooth development [77–79]. Since ACPs are presumed to emerge from the stomodeum, the detection of BMP4 within ACPs supports this hypothesis. In tooth development, BMP4 was shown to be capable of maintaining Wnt signalling by activating the expression of the LEF1 transcription factor [80], which is expressed in β-catenin accumulating ACP cells [19]. Both proteins are expressed in the enamel knot, which functions as a signalling centre in tooth development. Another enamel knot marker, p21WAF1/CIP1 [79], was also shown to be highly expressed in β-catenin accumulating tumour cell clusters (Fig. 5) [81, 82]. These observations substantiate the notion that ACPs express various tooth specific genes, exhibit an odontogenic morphological appearance [17, 19] and are likely to arise from oral ectodermal precursors. In colorectal cancer BMP4 expression was explicitly described in association with β-catenin mutations and enhanced Wnt signalling [83].
Fascin
Fascin was identified as a target gene of β-catenin in colon cancer [84]. Accordingly, the Fascin promoter and the first intron have consensus sequences matching with β-catenin interacting transcription factors of the TCF/LEF family [85]. Fascin, also known as Fascin-1, is a globular protein of 55 kDa that consists of 493 amino acids and acts as an actin bundling protein. It is expressed in vascular endothelial cells, fibroblasts, neuronal tissue and mature antigen presenting cells, but is predominantly absent in epithelial cells [86]. In humans, two additional paralogs of Fascin, Fascin-2 and Fascin-3, are expressed exclusively in the retina and the testis, respectively [86]. Fascin is involved in the migration machinery and reorganisation of the actin cytoskeleton. It cross-links filamentous actin (F-actin), which is formed by actin monomers that polymerise to fibres and coil with a second fibre. These structures are characterised by enhanced cytoskeleton rigidity and serve as scaffolds in cellular protrusions, the so-called filopodia. Filopodia formation plays a crucial role in cell migration, wound healing, neurite outgrowth, and serves as a precursor of dendritic spines in neurons [87]. The initiation and maintenance of filopodia formation is strongly regulated via extra- and intracellular signals. However, Fascin recently became a focus of interest, as aberrant expression was detected in various carcinomas, in particular at the invasion front of colorectal cancer and gastric cancer, where it has been identified as a target gene of β-catenin TCF signalling [84, 88]. Tumours with an enhanced Fascin expression were shown to be associated with increased invasive growth behaviour which resulted in an unfavourable prognosis for patients [89, 90].
Increased Fascin expression could be involved in sequestration and therefore restricted accumulation of β-catenin in ACP . The β-catenin protein binds Fascin by its armadillo repeat sequence which competes with E-cadherin and APC interaction (Fig. 4) [58, 59]. This suggests a cytoplasmic reallocation of β-catenin, since changes in cell adhesion, as well as a reduction of proteosomal destruction, occur [56].
Activated EGFR
Several studies showed that activated canonical Wnt signalling can be associated with an activated epidermal growth factor receptor (EGFR) cascade [91, 92] and vice versa [93], which is mediated by direct interactions between EGFR and β-catenin [94]. EGFR expression was shown to be increased in ACP and activated EGFR (phosphorylation of T1068) is detectable in the cytoplasm, as well as the nucleus of β-catenin accumulating cells (Fig. 5) [95]. The EGFR is a 170 kDa tyrosine kinase, also referred to as HER1, ErbB1, mENA and PIG61, which belongs to the human epidermal growth factor receptor (HER) family, comprising four members (HER1-4). As a transmembrane glycoprotein , it consists of an extracellular domain facilitating ligand binding, a transmembrane region and an intracellular region that comprises the tyrosine kinase domain and autophosphorylation sites (Fig. 6). Numerous growth factors bind to and activate the EGFR, including the epidermal growth factor (EGF), transforming growth factor alpha (TGF-alpha), amphiregulin, epiregulin, heparin-binding EGF (HB-EGF) and betacellulin [96, 97]. Ligand binding initiates a conformational change that causes receptor dimerisation and possesses protein-tyrosine kinase activity.
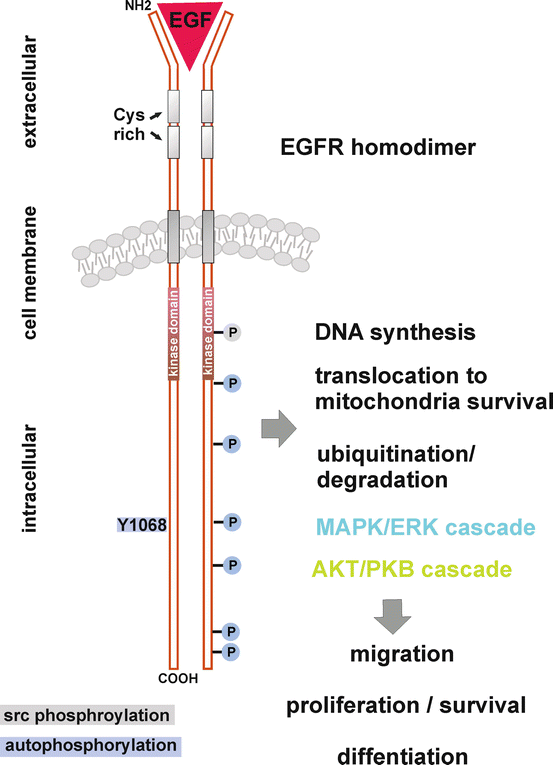
Fig. 6
Signal transduction of an activated EGFR. This illustration shows a subset of the most important tyrosine residues involved in EGFR signalling. Ligand binding to the extracellular region induces EGFR dimerisation. Intracytoplasmic tyrosine residues are autophosphorylated (blue) by its intrinsic tyrosine kinase, or phosphorylated by src kinase (grey). Adapter proteins bind to these phosphorylated tyrosine residues, thereby initiating the activation of diverse downstream cellular cascades, associated with cellular migration, proliferation, survival and differentiation. Adapted from www.cell-signalling.com
The EGFR protein-tyrosine kinase domain is composed of amino acid 686 to 960 which is encoded by exon 18 to 21 of the EGFR gene [96, 98]. The receptor dimer undergoes transphosphorylation on multiple tyrosine (Y) residues, which leads to the recruitment of a plethora of enzymes and adapter proteins acting as signal transducers and activators of at least five different intracellular cascades [99].
The major downstream signal transduction pathways activated by receptors of the HER family are the Ras-Raf mitogen-activated protein kinase (MAPK) pathway (MAPK/ERK cascade) and the phosphatidylinositol 3-kinase (PI3K)/Akt pathway (Fig. 6). Phospholipase Cγ (PLCγ), the signal transducer and activator of transcription (STAT) and src/FAK cascades are also stimulated [97, 99]. This signalling network possesses various cellular processes, e.g. gene expression, proliferation, inhibition of apoptosis, angiogenesis and cell migration [97].
The activation of the EGFR in an autonomous fashion through gene amplification or mutational events paves the way for human epithelial cancer development. The EGFR is overexpressed in the majority of solid tumours , including breast cancer, head and neck cancer, non-small cell lung cancer (NSCLC), renal cancer, ovarian cancer, and colon cancer and in a smaller percentage, also in bladder cancer, pancreatic cancer and gliomas [97]. EGFR overexpression produces an intense signal generation and activation of downstream signalling pathways (Fig. 6), resulting in a high rate of tumour growth, poor tumour differentiation and elevated invasive characteristics. In several lesions, mutations of the EGFR have been described in the tyrosine kinase domain, or mutations leading to truncation as a result of exons 2–7 deletion comprising amino acids 6–273 of the extracellular ligand domain [99], resulting in a ligand independent activation. This truncated EGFR variant, designated as EGFRvIII, is found in gliomas (38%) and NSCLC (5%) [97, 99–101]. Mutations within the tyrosine kinase domain are observed in 26% of NSCLC. Nearly 90% of alterations affect hotspots within exon 18–21 that code for tyrosine kinase. In frame deletions in exon 19, codons 746–750 account for 45–50% and substitution of leucine 858 for at least 35–45%. Insertion in exon 20 was found in 5% and substitutions spanning exon 18–21 occur rarely. Mutations exhibit oncogenic activities by selective activation of anti-apoptotic pathways through increased PI3K/AKT signalling [99].
In ACPs , DNA screening revealed that no activating mutations of the cytoplasmic EGFR autophosphorylation site encoded by exons 17–28 were detectable. The collection of normal controls showed a similar distribution of SNPs, suggesting that they are irrelevant in craniopharyngioma pathogenesis. Further examination of extracellular receptor regions excludes the occurrence of the EGFRvIII variant responsible for autonomous activation. In addition, no EGFR amplification was identified in ACP [95]. As non-genetic alterations are found, activation of the EGFR seems to occur in an autocrine fashion, by secreted soluble ligands or by components of its environment.
Indeed, EGF was found to be upregulated in β-catenin accumulating cell clusters in the embryonic mouse model of ACP and microarray gene expression studies of ACP identified increased amphiregulin levels, implying EGFR activation in an autocrine manner [102, 103]. Recent studies revealed that tenascin-C, a component of the extracellular matrix, is also capable of acting as an EGFR ligand, inducing its dimerisation and subsequent activation [104, 105]. It is worth nothing that ACPs establish a circumscribed tumour surrounding area, including explicit expression of the extracellular matrix protein tenascin-C at the invasion front (Fig. 5) [106]. However, additional studies are needed to determine whether this extracellular matrix protein corresponds to EGFR activation in ACP.
SHH
Several reports support a role of Sonic Hedgehog (SHH) signalling in the pathogenesis of ACP. No mutations in components of the SHH pathway could be identified in ACP [107]. However, there was one report describing a Patch 1 mutation leading to multiple Gorlin syndrome associated aberrations, e.g. basocellular tumours, bone and craniofacial malformations, and interestingly, even the occurrence of a craniopharyngioma. This mutation was observed to induce β-catenin accumulations in craniopharyngioma tissue [108].
HH signalling plays an important role during embryogenesis in cell differentiation and organ formation, e.g. in the brain, skin, intestine, teeth and pituitary gland [109–114]. Proper differentiation processes occur in a dose-dependent manner, pointing to a strict regulation of this pathway. In adults, SHH pathway is involved in stem cell maintenance and proliferation [115]. SHH as a paracrine acting factor is posttranscriptional modified by cholesterol maintaining a lipid anchor [113].
Extracellular SHH binds as a ligand to the transmembrane receptor Patched 1 (PTCH1), thereby, abrogating the inhibitory effect to smoothened (SMO), belonging to the frizzled class receptors, which subsequently induces downstream activation of the zinc finger proteins glioma-associated oncogene homologue 1–3 (Gli1–3) via the protein Fused (Fu). The activation of the Gli transcription factors could be inhibited by the proteinkinase A (PKA) and the Suppressor of fused (SuFu). The SHH inhibitory receptor Patch1 is a target gene of Gli transcription factors, and its upregulation represents a negative feedback loop [115].
A role of SHH pathway in ACP was first identified in the ACP mouse models [103]. In humans, ACP whorl like structures were shown to express SHH (Fig. 5) and Patch1 [102, 103]. Furthermore, Gli1, Gli3, SMO and SUFU were publicised not only as being expressed in the cytoplasm and within the nucleus in whorl configured cells, but also in the palisaded cell layer of ACP [108]. The SHH pathway seems to play mainly a relevant role in ACP pathogeneses, since comparative gene expression and methylation profiling revealed an increased expression of Gli2 and Ptch1 in ACP in comparison to PCP [7].
Impact of Activated Signalling Pathways on Proliferation and Differentiation in ACPs
Although the β-catenin-TCF pathway has a well-established role in activating cell proliferation processes [52], a decreased proliferation activity, as indicated by negative Ki67 staining [46, 81] and increased staining levels of the cell cycle inhibitor p21WAF1/CIP1, was shown [81, 116]. The proliferation activity correlating with the Ki-67 staining was shown to be low (<1% in high power microscopic inspection) or absent in epithelial whorls with β-catenin accumulations (Fig. 5). Furthermore, the distribution of p21WAF1/CIP1 is prominent in β-catenin accumulating whorls (Fig. 5). The p21WAF1/CIP1 immunoreactivity is also detectable beyond these cell clusters, e.g. in surrounding nests of ghost cells, although to a variable extent. In contrast, proliferating tumour cells are confined to the outer squamous epithelium, especially the basal cell layer [81]. During squamous differentiation of endometrial carcinoma cells, β-catenin is known to induce p21WAF1/CIP1 expression, thereby inhibiting cell proliferation [117]. A similar mechanism may play a role in ACP.
Related posts:
 Diagnosis of Human ACP
Diagnosis of Human ACP
 Deficits in Patients with Human Craniopharyngioma
Deficits in Patients with Human Craniopharyngioma
 Administration of Interferon-Alpha for Reduction of Cystic Tumour Burden
Administration of Interferon-Alpha for Reduction of Cystic Tumour Burden
 Management and Clinical Trials in Adamantinomatous Craniopharyngioma
Management and Clinical Trials in Adamantinomatous Craniopharyngioma
 Modified Mouse Models of Adamantinomatous Craniopharyngioma
Modified Mouse Models of Adamantinomatous Craniopharyngioma
 and Radiotherapy of Craniopharyngioma
and Radiotherapy of Craniopharyngioma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


