Fig. 1
Summary of the major mechanisms causing CIN in human cancer cells. The major defects leading to chromosome missegregation in human cancer cells are a premature loss of sister chromatid cohesion and the persistence of microtubule-kinetochore attachment errors that subsequently lead to the generation of lagging chromosomes. Various mechanisms can lead to these major causes of chromosome missegregation and the most important routes such as impaired chromatid cohesion, defects in SAC function, increased microtubule-kinetochore attachment stability, supernumerary centrosomes and increased microtubule plus end assembly are depicted
5.1 Defects in the Mitotic Spindle Assembly Checkpoint
It is conceivable that an impairment of the spindle assembly checkpoint (SAC) results in premature sister chromatid separation, which gives rise to chromosome missegregation and aneuploidy (Lara-Gonzalez et al. 2012; Musacchio 2011). In fact, this has been clearly demonstrated by deleting one allele of MAD2 in an otherwise chromosomally stable colon cancer cell line HCT116, which results in persistent chromosome missegregation and thus, is sufficient to cause CIN (Michel et al. 2001). Moreover, weakening the checkpoint in mice by partially reducing the expression of various SAC genes including MAD1, MAD2, BUB1, BUBR1, and BUB3 results in premature separation of sister chromatids, chromosome missegregation and CIN. Importantly, the majority of these mouse models demonstrated, for the first time, that the experimental induction of aneuploidy can support tumorigenesis and thus, verified Hansemann´s and Boveri´s hypothesis of a causal relationship of aneuploidy and tumorigenesis (Ricke et al. 2008; Pfau and Amon 2012). However, although inactivating mutations in BUB1 and MAD2, as well as reduced expression of MAD2 has been found in some cancer cell lines (Cahill et al. 1998; Li and Benezra 1996; Wang et al. 2002), large scale sequencing of human tumors has not revealed a frequent rate of SAC mutations in human tumors (Greenman et al. 2007; Wood et al. 2007; Jones et al. 2008; Parsons et al. 2008; Sjoblom et al. 2006). In addition, a functionally weakened SAC appears not to be a frequent event in cancer cells exhibiting CIN (Tighe et al. 2001). These findings indicate that impairments of the SAC can cause CIN by allowing chromosome segregation to occur in the presence of unaligned chromosomes, but are rarely detected in human cancer.
5.2 Sister Chromatid Cohesion Defects
Similar to an impairment of the SAC abnormal sister chromatid cohesion can also result in chromosome missegregation and CIN. Sister chromatids are held together by ring-shaped cohesin protein complexes, which consists of four different subunits including STAG1 or STAG2, RAD21, SMC1 and SMC3, which are loaded onto the chromosomes during DNA replication (Remeseiro and Losada 2013; Peters et al. 2008). In early mitosis the bulk of cohesin complexes dissociates from the chromosome arms while cohesion at the centromere is still maintained. The remaining cohesin complexes are released by cleavage of the RAD21 subunit by separase at the metaphase to anaphase transition. Dysfunction of any of the cohesin subunits can be expected to impair chromatid cohesion and may allow unscheduled separation of sister chromatids before full alignment on a metaphase plate occurs. In fact, mutations in genes encoding for cohesin subunits, in particular in STAG2, have been identified in various human cancers including bladder cancer, colorectal cancer, glioblastomas or myeloid neoplasms (Solomon et al. 2011, 2013; Welch et al. 2012). These mutations are indeed associated with improper chromatid cohesion and are sufficient to mediate CIN. Similarly, heterozygous knockout of STAG1 in mice also results in aneuploidy and supports tumorigenesis, but this effect might be mediated through incomplete telomere replication (Remeseiro et al. 2012). Since cohesin complexes also fulfill important functions outside of mitosis, e.g., for gene transcription (Remeseiro et al. 2013) it remains possible that cancer-associated mutations of cohesin genes might drive tumorigenesis through mechanisms independent of premature sister chromatid separation during mitosis.
5.3 Erroneous Microtubule-Kinetochore Attachments and Lagging Chromosomes
Albeit detectable, premature separation of sister chromatids occurs in human cancer at rather low rate. Thus, other mechanisms must be responsible for widespread chromosome missegregation events as seen in cancer cells. The most common phenotype observed in mitotic cancer cells and leading to chromosome missegregation is the presence of so-called lagging chromosomes during anaphase (Thompson and Compton 2008). Lagging chromosomes are the result of erroneous microtubule-kinetochore attachments that are not resolved before anaphase onset (Cimini 2008). Since the capture of kinetochores by microtubules during the early phases of mitosis is a largely stochastic process, it is conceivable that kinetochore mal-attachments occur at a regular basis. Those mal-attachments include mono-, syn-, and merotelic attachments that need to be corrected, in order to establish proper amphitelic attachments that are a prerequisite for proper chromosome alignment and segregation (Fig. 2). In contrast to mono- and syntelic attachments, merotelic attachments, characterized by kinetochores being attached to microtubules emanating from the two opposite poles at the same time, represents a particular problem for a mitotic cell. These attachments fully occupy the kinetochores and are not recognized by the SAC, thus allowing the onset of anaphase despite the presence of this erroneous kinetochore attachment (Cimini 2008). This results in one (or more) lagging sister chromatids(s) that cannot properly be segregated towards one pole of the cell and thereby, creates a pre-stage of chromatid missegregation (Fig. 2). Intriguingly, the presence of lagging chromosomes is most prevalent in cancer cells exhibiting CIN and it is assumed that this represents a major route to chromosome missegregation and aneuploidy in cancer (Thompson and Compton 2008; Cimini et al. 2001). However, the molecular mechanisms leading to the generation of erroneous kinetochore attachments might be various and are discussed below.
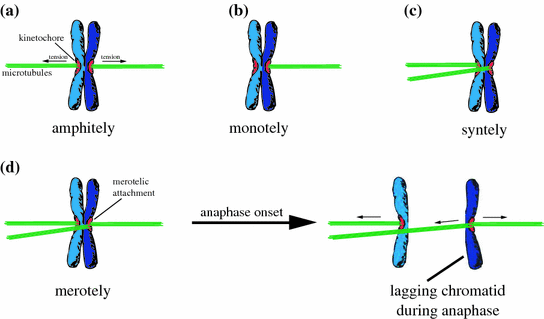
Fig. 2
Schematic depiction of erroneous microtubule-kinetochore attachments. a Amphitelic attachment. Both sister kinetochores are correctly attached in a bi-oriented manner to microtubules emating from the two opposite centrosomes. b Monotelic attachment. Only one sister kinetochore is attached to microtubules from one centrosome. c Syntelic attachment. Both sister kinetochores are attached to microtubules emanating from the same pole. d Merotelic attachment. Both sister kinetochores are attached to microtubules from the two opposite poles, but one kinetochore is bound to microtubules from both poles. After anaphase onset merotelic attachments cause the generation of a lagging chromatid, which cannot be properly segregated
5.4 Abnormal Microtubule-Kinetochore Attachment Stability
The interaction of microtubules with kinetochores during the early phases of mitosis is a highly dynamic process, during which finally around 20 microtubules are bound to one kinetochore. Erroneous attachments are resolved by de-stabilization of these interactions while correct attachments are stabilized. The cycles of attachment, release and re-attachment are called error correction and are required for achieving bi-oriented amphitelic kinetochore attachments for every chromosome (Nicklas and Ward 1994). The dynamic turnover of microtubules bound to kinetochores (so-called k-fibres) can be measured in live cells by determining fluorescence dissipation after photo-activation of PA-GFP-tubulin within mitotic spindles. These measurements revealed that the turnover of k-fibres is high in early mitosis (t 1/2 = 2–3 min) and low in metaphase (t 1/2 = 6–7 min) indicating that error correction is more active in early mitosis than in cells that have achieved full chromosome alignment (Kabeche and Compton 2013; Zhai et al. 1995). It is obvious that error correction takes place at the kinetochore-microtubule interface and therefore, kinetochore-based microtubule depolymerases are key to error correction. In fact, microtubule depolymerases of the kinesin-13 family such as MCAK or Kif2B are directly involved in destabilizing erroneous kinetochore attachments, thereby releasing faulty attached kinetochores from the microtubule plus ends (Bakhoum et al. 2009a, b; Maney et al. 1998). In addition, other kinetochore proteins such as the Hec1-Ndc80 complex, which is frequently deregulated in human cancer, also contribute to the stabilization of the k-fibres (DeLuca et al. 2006). Most notably, the Aurora-B kinase, which localize to the inner centromere can directly regulate the localization and activity of MCAK and Kif2b and can thereby contribute to destabilization of k-fibres and to error correction (Cimini et al. 2006; Hauf et al. 2003; Knowlton et al. 2006; Andrews et al. 2004). Consequently, inhibition of Aurora-B or loss of Kif2b or MCAK causes hyper-stability of microtubule-kinetochore attachments leading to lagging chromosomes and chromosome missegregation. How a cell recognizes erroneous kinetochore attachments and how it can discriminate between proper and improper attachment is currently not entirely clear, but might be related to the fact that only proper amphitelic attachments are able to provide the basis for the generation of full tension across the two sister kinetochores. In this situation, the inner centromere-based Aurora-B kinase might be spatially separated from their substrates, namely the kinetochore-based microtubule depolymerases, which can then no longer de-stabilize the microtubule-kinetochore attachments (Liu et al. 2009).
Most intriguingly, hyper-stable microtubule-kinetochore attachments are detected in many cancer cells exhibiting CIN suggesting that erroneous kinetochore attachments cannot be properly resolved in those cancer cells (Bakhoum et al. 2009a, b). In fact, reduced turnover of microtubules bound to kinetochores correlates with the subsequent appearance of lagging chromosomes during anaphase and with an increased rate in chromosome missegregation in human cancer cell lines. Moreover, overexpression of KIF2B or MCAK results in de-stabilization of these hyper-stable attachments and is sufficient to suppress chromosome missegregation in cancer cells with CIN (Bakhoum et al. 2009b). These findings suggest that cancer cells might harbor an insufficiency of the error correction machinery, but so far no frequent alterations in genes known to play a role in error correction have been found. Genes such as AURORA–B, KIF2B or MCAK are found rarely or not at all mutated or inactivated in human cancer (Greenman et al. 2007; Wood et al. 2007; Jones et al. 2008; Parsons et al. 2008; Sjoblom et al. 2006). Nevertheless, other genes not necessarily directly involved in error correction might contribute to hyper-stable kinetochore attachments. An important example might be the tumor suppressor gene APC, which is frequently inactivated in human cancer and which was previously shown to be involved in mediating the interaction of microtubule plus tips with kinetochores (Fodde et al. 2001). Loss of APC results in hyper-stable kinetochore attachments, lagging chromosomes and whole chromosome missegregation, but this might also reflect the generation of erroneous kinetochore attachments per se (Bakhoum et al. 2009a). Similarly, overexpression of the SAC gene MAD2 was found to be frequent in human cancer and instead of mediating hyper-activity of the mitotic spindle checkpoint, it was found to cause hyper-stable kinetochore attachments resulting in lagging chromosomes and aneuploidy (Kabeche and Compton 2012). Whether this also applies to other SAC genes remains to be seen. Interestingly, a weakened spindle checkpoint can also be associated with the generation of lagging chromosomes. The molecular basis for this is still unclear, but one could argue that cells without a proper SAC might have a reduced time window for error correction before anaphase is initiated and thus, uncorrected kinetochore attachments can persist leading to the appearance of lagging chromosomes during anaphase.
5.5 Supernumerary Centrosomes
Given the fact that the presence of lagging chromosomes is a major phenotype of human cancer cells exhibiting CIN it is important to emphasize that there are at least two ways to achieve this important route to chromosome missegregation: (i) reduced error correction as discussed above leading to the persistence of unresolved erroneous kinetochore attachments. (ii) increased rate of the generation of erroneous kinetochore attachments that might simply overwhelm the cellular (in principle functional) error correction machinery.
As shown recently, the presence of supernumerary centrosomes is one important way to increase the rate of erroneous kinetochore attachments (Ganem et al. 2009; Silkworth et al. 2009). Since the days of Boveri, it is well known that cancer cells frequently exhibit more than two centrosomes. In fact, about 20–30 % of cancer cells might contain more than two centrosomes (Ghadimi et al. 2000). Moreover, as Boveri already realized supernumerary centrosomes can be a source for chromosome missegregation and it was assumed for many years that supernumerary centrosomes can give rise to multipolar mitotic spindles, which inevitably would result in highly unequal chromosome segregation. Indeed, multipolar spindles can be seen occasionally in cancer cells with supernumerary centrosomes, which further supported this assumption. However, Pellman, Cimini and collegues followed the fate of cancer cells after undergoing multipolar mitoses and it became clear that multipolar chromosome missegregation is not compatible with cell viability (Ganem et al. 2009; Silkworth et al. 2009). Instead, the majority of cells containing supernumerary centrosomes only transiently build up a multipolar spindle, which subsequently is re-organized into a bipolar spindle by clustering the supernumerary centrosomes together into two poles. In the end, most cancer cells with supernumerary centrosomes undergo bipolar mitoses, albeit with reduced fidelity. Interestingly, the transient formation of multipolar spindle intermediates followed by centrosomes clustering facilitates the generation of erroneous kinetochore attachments and consequently, leads to lagging chromosomes. Thus, centrosome clustering provides a plausible mechanism that explains the survival of cancer cells in the presence of more than two centrosomes. The transient spindle geometry abnormality arising from this explains the strong correlation between supernumerary centrosomes and CIN in human cancer (Ganem et al. 2009; Silkworth et al. 2009). Although a number of genes have been identified that are required for centrosome clustering in human cancer cells (Kwon et al. 2008) the detailed mechanism of how centrosome clustering is regulated is currently not well understood.
5.6 Increased Microtubule Plus End Assembly
Most recently, an increase in microtubule plus end assembly rates within mitotic spindles has been identified as a key trigger for CIN in human cancer cells (Ertych et al. 2014). In fact, this increase in microtubule plus end dynamics is sufficient to cause transient spindle geometry abnormalities that facilitate the generation of kinetochore mal-attachments in the presence of a fully functional error correction machinery. Importantly, restoration of proper microtubule plus end assembly rates cannot only suppress the transient spindle abnormalities and the generation of lagging chromosomes, but also restores chromosomal stability in otherwise chromosomally instable human cancer cells. This clearly establishes a causal relationship between increased spindle microtubule dynamics, lagging chromosomes, and CIN. Moreover, this particular phenotype appears to be highly frequent in human cancer and can be mediated by cancer-relevant genetic lesions such as amplification of AURKA [encoding for the centrosomal Aurora-A kinase; (Marumoto et al. 2005)] or the loss of the tumor suppressor genes CHK2 and BRCA1, which have been previous implicated in the regulation of mitosis and for the maintenance of chromosomal stability (Stolz et al. 2010, 2011). However, the molecular mechanisms underlying the detected increase in microtubule plus end dynamics in cancer cells are still elusive. Nevertheless, the identification of this widespread mechanism leading to CIN further supports the notion that an increase in the rate of the generation of erroneous kinetochore attachments might overwhelm the cellular error correction capacity and thus, causes the persistence of lagging chromosomes during anaphase, finally leading to increased rates of chromosome missegregation (Ertych et al. 2014).
6 Abnormalities During Interphase Causing CIN
In addition to defects that arise during mitosis and directly causing chromosome missegregation it is conceivable that mechanisms that originate during interphase might also (indirectly) lead to chromosome missegregation. Some examples will be discussed in the next paragraphs.
6.1 Centrosome Amplification
As discussed, the presence of supernumerary centrosomes during mitosis causes transient spindle geometry abnormalities that facilitate the generation of erroneous microtubule-kinetochore attachments leading to chromosome missegregation. While the direct consequence of supernumerary centrosomes can clearly be seen during mitosis, supernumerary centrosomes originate from defects in the centrosome duplication cycle taking place during interphase (Meraldi and Nigg 2002). Centrosomes must duplicate only once per cell cycle and this process is initiated by centriole duplication at the G1 to S phase transition concomitant to DNA replication. Several proteins including Cep192, HsSAS-6 and PLK4 among others are required for proper centriole duplication. Conceivably, over expression of these key drivers of centriole duplication results in centrosome overduplication and can be indeed found in human cancer (Anderhub et al. 2012). Similarly, the CDK2-cyclin E kinase activity is also involved in centrosome duplication and overexpression of cyclin E is sufficient to induce centrosome amplification (Hinchcliffe et al. 1999). Increased cyclin E levels are detected in human cancer cells and can be induced either by amplification of the CYCLIN–E gene locus or by inhibition of the ubiquitin-proteasome dependent proteolysis of cyclin E (Rajagopalan et al. 2004; Spruck et al. 1999). In fact, the latter requires a substrate recognition subunit of the SCF ubiquitin ligase called hCDC4/Fbw7, which is frequently mutated in human cancer leading to an unscheduled accumulation of cyclin E. Consequently, loss of hCDC4/Fbw7 in human tumors is associated with centrosome amplification and aneuploidy (Rajagopalan et al. 2004).
6.2 Alterations in Gene Transcription Affecting Mitosis
In human cancer, there are numerous examples of transcription factors that are deregulated. So far, the best studied examples include oncogenic transcription factors such as c-myc, c-jun, or c-fos or the tumor suppressor protein p53 that mainly contribute to a de-regulation of the G1 to S phase transition of the cell cycle. However, there are also many examples of de-regulated gene transcription that directly affects genes involved in mitotic processes. For example, the transcription of MAD2 during G2 phase of the cell cycle is restrained by the repressor-element-1-silencing transcription factor (REST; also known as: neuron-restrictive silencing factor, NRSF) that must be degraded during G2 by the ubiquitin-proteasome pathway in order to allow the expression of proper levels of MAD2 (Guardavaccaro et al. 2008). However, REST is frequently over expressed in, e.g., neuro- and medulloblastomas (Su et al. 2006) leading to reduced expression of MAD2 resulting in an impairment of the checkpoint (Guardavaccaro et al. 2008). As discussed before, this can directly trigger chromosome missegregation in the presence of unaligned chromosomes.
MAD2 expression is also under control of the E2F transcription factors, which are negatively regulated by the retinoblastoma tumor suppressor protein (pRb). In fact, loss of RB, which is frequent in various types of cancer and associated with aneuploidy in tumors, can result in MAD2 overexpression (Hernando et al. 2004), which, as discussed already above, can contribute to the generation of hyper-stable microtubule-kinetochore attachments leading to lagging chromosomes and missegregation (Kabeche and Compton 2012). In addition, loss of RB has also been implicated in altering the centromere geometry, thereby facilitating microtubule-kinetochore attachment errors and aneuploidy (Manning et al. 2010).
Similarly, the forkhead transcription factor FoxM1 is required for proper progression of mitosis and for the maintenance of chromosomal stability by regulating the expression of various key regulators of mitosis including Plk1, cyclin B, and Cenp-F. (Laoukili et al. 2005). Importantly, FoxM1 is frequently overexpressed in human cancer resulting in overexpression of several mitotic regulators during mitosis (Fu et al. 2008). However, the molecular consequences of elevated levels of, e.g., cyclin B during mitosis and on chromosome segregation are currently not clear.
6.3 Replication Stress as a Source for CIN
Most recently, impaired replication fork progression and replication stress during S phase has been implicated as a source for chromosome missegregation during mitosis (Burrell et al. 2013). In fact, it was shown that several genes located on chromosome 18q, which is subject to frequent loss in cancer cells exhibiting CIN, can act as CIN suppressor genes. Moreover, loss of these CIN suppressor genes results in replication stress leading to the generation of acentric chromosome fragments that are missegregated during mitosis. Importantly, the missegregation of chromosome fragments after replication stress might also cause structural chromosome abnormalities frequently found in aneuploid cancer cells. However, whether this mechanism can also account for whole chromosome missegregation is under debate (Bakhoum et al. 2014).
Related posts:
 Modelling: What Have Mouse Models for Chromosome Instability Taught Us?
Modelling: What Have Mouse Models for Chromosome Instability Taught Us?
 of Aneuploidy in Cancer: Transcriptome and Beyond
of Aneuploidy in Cancer: Transcriptome and Beyond
 Diverse Effects of Complex Chromosome Rearrangements and Chromothripsis in Cancer Development
Diverse Effects of Complex Chromosome Rearrangements and Chromothripsis in Cancer Development
 of Chromosomal Aberrations in Solid Tumors
of Chromosomal Aberrations in Solid Tumors
 Dysfunction, Chromosomal Instability and Cancer
Dysfunction, Chromosomal Instability and Cancer
 as Models of Mitotic Fidelity
as Models of Mitotic Fidelity
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


