Fig. 1
Schematic illustration of commonly observed numerical and structural chromosome alterations identified in solid tumors, and the methodologies capable of their detection. Detection of an alteration, however, is not necessarily synonymous with the ability to unambiguously determine the genomic origin of aberrant material. Chromosome banding, SKY/M-FISH and chromosome comparative genomic hybridization (CGH) are low resolution techniques, whereas array-based CGH (aCGH), single nucleotide polymorphism (SNP) arrays and next-generation sequencing (NGS) are much higher resolution methodologies. (Adapted from Albertson et al. 2003)
2.1 Identification of Chromosome Translocations
The forty-six human chromosomes were initially organized into a karyotype based solely on their size and the positioning of their centromere, to which the mitotic machinery attaches. In 1968, Caspersson and his colleagues developed a method for staining chromosomes with quinacrine mustard, which resulted in a banding pattern (Q-bands) that was unique to each chromosome pair (Caspersson et al. 1968). Shortly thereafter, a similar methodology was developed involving the treatment of the metaphase chromosome preparations with the enzyme trypsin followed by staining with Giemsa. The binding of this stain to A-T base pairs resulted in patterns of alternating “light” and “dark” bands, which became known as G-bands (Seabright 1971; Sumner et al. 1971). While this technique was advantageous for unequivocally identifying and organizing normal chromosomes, it proved to be an enormous advance in terms of demonstrating the complexity of the cancer genome. Cancer karyotypes usually show complex rearrangements, involving genomic regions of different chromosomal origin combining in the formation of derivative chromosomes. It remained extremely difficult, however, for cytogeneticists to determine the exact composition of these “marker” chromosomes based solely on G-banding.
In the late nineteen-nineties, the development of two sophisticated multi-colored fluorescence in situ hybridization (M-FISH) methods utilizing a combination of whole-chromosome painting probes for uniquely labeling each chromosome pair, i.e., spectral karyotyping (SKY) and M-FISH, represented an enormous leap forward in our understanding of the underlying complexity of cancer karyotypes (Schrock et al. 1996; Speicher et al. 1996). Although the resolution was limited to the level of an individual chromosome band, these techniques allowed the assessment of specific chromosome partners involved in both balanced and unbalanced translocations, as well as the visualization of previously unidentified cryptic aberrations (Veldman et al. 1997). Nevertheless, the requirement for high-quality chromosome metaphase spreads from primary solid tumors remained a challenge, especially for tumors that are difficult to culture or have a relatively slow rate of cell division.
The utilization of both chromosomal banding-based techniques and SKY/M-FISH resulted in a plethora of well-annotated karyotypes for most cancer types archived in the Mitelman Database of Chromosome Aberrations in Cancer (http://cgap.nci.nih.gov/Chromosomes/Mitelman). It was not until the post-genome era, with the introduction of the next-generation sequencing technology, which allowed the in silico alignment of paired short reads from the ends of fragments covering the whole genome, that genomic rearrangements, including balanced translocations or inversions, could be defined at the individual nucleotide level.
2.2 Identification of Copy Number Changes
In 1992, Kallioniemi and colleagues introduced a genome-wide screening technique, termed CGH, which allowed visualization of chromosomal imbalances without the need to prepare tumor metaphase chromosomes (Kallioniemi et al. 1992). Total genomic DNA isolated from the patient’s tumor (i.e., test DNA) and from any other source of non-tumor tissue (i.e., reference DNA) were each labeled with a different fluorescent molecule, or fluorochrome. Equal amounts of labeled test and reference DNA were mixed and hybridized to normal lymphocyte metaphase chromosomes, which cytogeneticists were already capable of preparing for high resolution banding (Yunis and Chandler 1978). Deviations from a 1:1 intensity ratio between the two fluorochromes along the length of each chromosome indicated the gain or loss of genomic material in the tumor sample relative to a normal non-tumorous reference sample. Because this technique still relied on the usage of metaphase chromosomes, the resolution remained limited to the size of an individual band, or about 5–10 Mb (Carter 2007). Later, metaphase chromosomes were replaced by increasingly shorter normal genomic DNA fragments, including YACs, BACs, or oligonucleotides representing the entire genome spotted onto glass slides (Pinkel et al. 1998; Solinas-Toldo et al. 1997). This array-based CGH, or aCGH, resulted in a much higher resolution and flexibility, limited only by the size and spacing of the DNA fragments that were arrayed. Using an automated calculation of the ratio between the intensities of the two fluorochromes for each spotted feature on the microarray, a dedicated software provides a detailed map of genomic gains and losses distributed across the genome. In contrast to conventional CGH, the analysis of aCGH data does not require previous knowledge to identify chromosome pairs based on G-banding, which makes this methodology much more universal and powerful. Further developments in the sensitivity of array technology resulted in the ability to detect differences in the hybridization efficiency of two DNA fragments that differed in a single nucleotide (Carter et al. 2012). In addition to copy number changes, these single-nucleotide polymorphism or SNP-based arrays were capable of providing information regarding the haplotype of each allele. If an individual has different alleles at a particular genomic locus, SNP arrays provide the opportunity to determine if one allele is preferentially lost in the tumor. In addition, such a LOH in the presence of two copies of the locus or chromosome enables the detection of somatically acquired uniparental disomies (Tuna et al. 2009). Thus, the extent to which one platform is more appropriate than another depends on the study design and the type of information one is looking for.
Undoubtedly, the major advantage of CGH and SNP arrays is the amenability of any cancer specimen to DNA extraction. There are, however, some caveats that must be considered when using these methodologies. First, some 60–70 % of tumor purity is strongly recommended to be able to identify single-copy genomic alterations, i.e., “contamination” with normal, non-cancerous cells is highly problematic. Second, these methodologies only take a snapshot of the tumor lifespan, so intrinsic tumor heterogeneity may potentially dilute out the intensity ratio of the main clonal population. Hence, the detection of low-level copy number events may be limited due to the natural presence of subpopulations with different DNA content. Third, the amount of tissue available from fresh tumor samples, particularly in the case of biopsies, is often limited. It would therefore be advantageous to be able to assess copy number alterations by extracting DNA from formalin-fixed, paraffin-embedded (FFPE) tissue sections once histopathology has been performed. This remains challenging because of the low yield and poor integrity of the extracted DNA. Currently, very few groups have been able to successfully perform aCGH using a small number of cells isolated by microdissection from archival FFPE tissues sections (Al-Mulla 2011; Hirsch et al. 2012; Johnson et al. 2006; van Essen and Ylstra 2012) or from individual circulating tumor cells (Heitzer et al. 2013).
Recent incorporation of next-generation sequencing-based approaches led to the development of tools to infer copy number changes from whole-exome or whole-genome sequencing in a pretty reliable manner (Kendall and Krasnitz 2014). Compared with microarray experiments, these technologies not only allow the identification of alterations at the nucleotide level, but they also have the advantage that the signal intensity does not reach a point of saturation, thus they have a much higher dynamic range and a higher rate of detecting aberrations. High-coverage genome sequencing has been applied to detect clonal subpopulations to an unprecedented level (Gerlinger et al. 2014). Nevertheless, as the number of studies applying next-generation sequencing to large cohorts is limited by the costs, aCGH or SNP arrays remain the gold-standard methodology to assess copy number changes in solid tumors.
3 Structural Chromosomal Rearrangements in Solid Tumors
The study of chromosomal abnormalities in cancer underwent a paradigm shift with the discovery of the Philadelphia chromosome in patients with chronic myelogenous leukemia (CML) by Nowell and Hungerford (1960). The genomic composition of this aberrant chromosome was later determined by Janet Rowley to result from a balanced translocation between chromosomes 9 and 22, or t(9;22) (Rowley 1973). The staining of chromosomal preparations of cells at the metaphase stage with Giemsa was utilized to identify the t(9;22). This landmark discovery initiated the description of marker chromosomes in a plethora of human cancers. In leukemia and lymphoma, the application of this technique to identify aberrant chromosomes has led to improved treatment and clinical outcomes for many patients, and it is still being used for clinical assessment (Rampal and Levine 2013; Rowley 2008).
While chromosome translocations, inversions, and insertions are typically observed in cancer (Albertson et al. 2003), balanced translocations, in which material from both partner chromosomes is retained by the cell, are often identified in hematological malignancies (e.g., t(9;22) in CML or t(8;14) in Burkitt’s lymphoma). The most likely explanation is that site-specific DNA recombination of antigen receptor genes is an essential physiological step for the development of mature B- and T-lymphocytes. Any error in the regulation of this process could result in the rearrangement of other genomic regions. Juxtaposition of a cellular proto-oncogene to an actively transcribed region of the genome has the potential to generate a cell with a growth and/or survival advantage. Thus, these aberrations tend to be causal in the development of hematological malignancies, and therefore drugs designed to target the resulting proteins have proven to be extremely effective.
In contrast, partial deletions, duplications, and unbalanced translocations (i.e., rearrangements in which genomic material is lost) are the most frequent chromosomal alterations identified in cancers of epithelial origin (Mitelman et al. 1997). Distinct patterns of recurrent chromosomal translocations in these tumors are extremely rare (Mitelman 2000). One explanation could be the difficulty of identifying and mapping structural rearrangements in these karyotypically complex tumors. Although this issue is currently overcome by the usage of next-generation sequencing approaches, newly generated data have failed to demonstrate the relevance of such rearrangements in the development of epithelial cancers. Another reason for the rare occurrence of recurrent structural chromosome aberrations in carcinomas could be tissue-specific differences in the mechanisms responsible for their generation, such as the absence of site-specific recombination in non-lymphocytic cells.
The identification of recurrent translocations has only been described in a few solid tumor types. A number of approaches, including cancer outlier profiling analysis of gene expression signatures (Tomlins et al. 2005) and next-generation RNA and DNA sequencing, have demonstrated the presence of gene fusions that result in altered transcriptional expression or protein activity in some of the main epithelial cancer types, although at a frequency of only about 10 % (Mitelman et al. 2004, 2005, 2007; Giacomini et al. 2013). Prostate cancer (TMPRSS2–ETV1, TMPRSS2–ETV4, TMPRSS2–ETV5) (Kumar-Sinha et al. 2008), colorectal cancer (VTI1A–TCF7L2, NAV2–TCF7L1) (Bass et al. 2011; Cancer Genome Atlas Network 2012a), papillary thyroid carcinoma (RET–NTRK1) (Wells and Santoro 2009), papillary renal cell carcinoma (PRCC–TFE3) (Kauffman et al. 2014), and non-small-cell lung cancer (NSCLC) (RET and ROS1) (Oxnard et al. 2013; Shames and Wistuba 2014) are examples of solid tumors with chromosomal rearrangements generating gene fusions with biological and, potentially, clinical implications.
4 Cancer Ploidy and Chromosome Aberration Rates
Solid tumors with a chromosome number between triploid (n = 69) and tetraploid (n = 92) have been estimated to occur in some 30 % of all epithelial cancers (Storchova and Kuffer 2008). As it is unlikely that chromosome missegregation alone occurs at sufficiently high rates to explain how cancer cells achieve such pseudo-polyploid karyotypes during the tumor lifespan, it has been proposed that the cancer genome first undergoes a whole genome duplication event. This tetraploidization, being an unnatural event, is thought to be highly unstable, allowing for the development of structural abnormalities and selective loss of chromosomes until the genome somehow becomes stable again (Burrell et al. 2013). Tetraploidy and high levels of aneuploidy are often correlated with disease aggressiveness, poor prognosis and the generation of metastases (Camps et al. 2004; Gerlinger et al. 2012). In fact, ongoing rates of chromosome missegregation events define levels of genomic instability in several cancer types (Camps et al. 2005). In colorectal cancer (CRC), for instance, the rate of chromosomal instability is directly related to the mutational status of genes involved in the DNA mismatch repair pathway (Lengauer et al. 1998). Near-diploid colorectal tumors are mismatch repair deficient, whereas aneuploid tumors contain intact repair pathways and show higher rates of both numerical and structural chromosome alterations, features observed in the majority of human carcinomas (Lengauer et al. 1997). In the past, the only plausible strategy to identify the total number of chromosomes per cell was to prepare and analyze metaphase chromosomes from dividing cells. Recently, application of SNP arrays in combination with specific analytical tools attempts to further define absolute allelic copy number changes allowing the determination of the tumor ploidy directly from tumor tissue (Van Loo et al. 2010; Carter et al. 2012).
5 Recurrent Low-Level Copy Number Alterations Among Different Cancer Types: Defining the Cancer Genome
Aneuploidy represents a ubiquitous feature of cancer cells of epithelial origin, and usually implies growth advantages, poor prognostication and shortened patient survival (Gordon et al. 2012; Holland and Cleveland 2009); therefore, gains and losses of chromosomes are positively selected throughout the tumor lifespan. As a result, most cancer genomes show a modal chromosome number far from the normal diploid genome of 46 chromosomes. Low-level copy number changes usually include genomic imbalances that affect the entire chromosome or a chromosome arm, regardless of parameters such as size or gene density. The identification of low-level copy number alterations by karyotyping, CGH and next-generation sequencing provides supporting evidence of a distinct pattern of genomic imbalances depending on the tumor’s tissue of origin. In this section, we will describe some examples of the tumor-type specific distribution of copy number alterations.
CRC, being among the more amenable solid tumors to cytogenetic analyses, is one of the most well-studied cancer genomes. Bardi and colleagues systematically cultured colon cancer cells from primary specimens and reported extensive cytogenetic data on both the tumors and derived cell lines, plotting the results as chromosome maps of gains and losses (Bardi et al. 1993, 1995). Later, using conventional CGH, Ried and his colleagues described recurrent alterations in sporadic (i.e., non-hereditary) CRCs in which genomic gains affecting chromosomes 7, 8q, 13, and 20q occurred with frequencies upwards of 80 %, and genomic losses of chromosomes 4, 8p, 17p, and 18q were often observed (Ried et al. 1996). In addition, several reports have shown that some of these aberrations, mainly the gain of 7 and 20q, can already be observed in preneoplastic polyps (Habermann et al. 2011), and most, if not all, are still present in liver metastases of this disease and in in vitro models derived from primary tumors or metastasis (Camps et al. 2009; Platzer et al. 2002). The plethora of conventional and array-based CGH studies applied to map genomic imbalances in CRC convincingly confirmed these earlier results [reviewed in (Grade et al. 2006a)], supporting the idea of a genomic ID associated with CRC. These chromosomal aberrations, as highlighted in Fig. 2, actually accompany the genetic (mutational) and epigenetic events comprehensively described in The Cancer Genome Atlas (Cancer Genome Atlas Network 2012a), and serve as the basis for the CRC progression model published by Bert Vogelstein and Eric Fearon more than two decades ago (Fearon and Vogelstein 1990; Vogelstein et al. 1988).
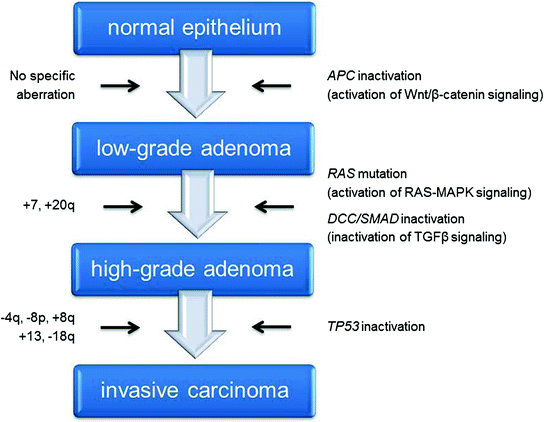
Fig. 2
Progression model of colorectal carcinogenesis. The progression of low-grade adenomas to high-grade adenomas is accompanied by gains of chromosomes 7 and 20q. Gains of chromosomes 8q and 13, as well as losses of chromosomes 4p, 8p and 18q, indicate transition into invasive carcinomas. These chromosomal aberrations, which are specific for colorectal cancer, accompany the genetic (mutational) changes observed at the level of individual genes that serve as the basis for the colorectal cancer progression model, referred to as the adenoma-carcinoma-sequence by Vogelstein and Fearon
It is interesting to note the high tissue-specificity of some of the genomic imbalances described above. For example, the gain of chromosome 13 is practically exclusive to colorectal neoplasms. Although it is not one of the earliest chromosome aneuploidies observed in adenomas, the incidence reaches some 70 % in carcinomas and metastases. The presence of several driver genes (i.e., CDK8, CDX2, LNX2, and DIS3), relevant for colorectal carcinogenesis and distributed along the length of this chromosome, favors the gain of the whole chromosome over a short focal amplification, and defines chromosome 13 as a CRC chromosome (Camps et al. 2013; de Groen et al. 2014; Firestein et al. 2008; Salari et al. 2012). Although not as exclusive as the gain of chromosome 13, the gain of chromosome arm 20q occurs in more than 60 % of CRC. Based on integrative genomic approaches, multiple putative oncogenes could drive the selection of this region of the genome in colorectal and other cancer types (Carvalho et al. 2009). Notably, rectal cancers exhibit mirroring genomic profiles compared to colon cancers (Grade et al. 2006b, 2009).
Additionally, besides specific focal events, similar patterns of gross genomic imbalances are observed in other gastrointestinal adenocarcinomas such as esophageal and gastric cancers (Dulak et al. 2012). In contrast, non-small cell lung carcinoma, prostate cancer, ovarian cancer, breast cancer, glioma and others display a loss of chromosome 13 (Di Fiore et al. 2013), as it also contains the well-known tumor suppressor gene RB1 at 13q14, thus demonstrating that the mechanisms to instigate carcinogenesis depend on different driver genes in a tissue-of-origin specific fashion.
The fact that chromosomal gains and losses define specific tumor entities also applies to breast cancer, even though the picture is a bit more complex because of the heterogeneity of this disease (Kallioniemi et al. 1994; Pollack et al. 2002; Ried et al. 1995). With higher resolution CGH techniques, primary breast carcinomas could be discerned into three groups: (i) near-diploid tumors characterized by extra copies of chromosome arm 1q and losses of 16q, thus referred to as “1q/16q” tumors; (ii) aneuploid tumors defined by recurrent copy number gains of 8q and extensive chromosomal instability, named “complex”; and (iii) aneuploid tumors with frequent focal high-level amplifications, e.g., of the oncogenes CCND1, MYC, and ERBB2, also known as the “amplifier” group (Fridlyand et al. 2006). Integration of gene expression signatures defined five major breast cancer subtypes (basal-like, luminal A, luminal B, ERBB2, and normal breast-like) (Perou et al. 2000), and copy number alteration data showed that recurrent genomic aberrations differ between these subtypes and that stratification correlated with clinical outcome (Chin et al. 2006). In addition, recent studies that applied quantitative measurements of the nuclear DNA content to primary breast carcinomas unambiguously established that tumors with a higher degree of chromosome instability were associated with a worse prognosis (Habermann et al. 2009). Of note, the genomic profiling of serous ovarian cancer partially resembles that of basal-like breast carcinomas, including gains of 1q, 3q, 5p, and 8q, and losses of chromosome 4, 5q, 8p, and 13 (Cancer Genome Atlas Network 2012b; Cancer Genome Atlas Research Network 2013).
In a completely different cancer lineage, the application of molecular cytogenetic techniques also revealed that essentially all cervical carcinomas exhibit an extra copy of the long arm of chromosome 3 (Heselmeyer et al. 1996). The gain of 3q is already present in dysplastic precursors, and, in fact, the presence of this single cytogenetic aberration discerns those lesions that will eventually progress from those that will not (Heselmeyer et al. 1997). Deduced primarily from retrospective studies of Pap smears using interphase FISH with a series of probes for the enumeration of 3p, 3q and cellular ploidy, the gain of 3q determines the acquisition of invasiveness capacities, thereby demonstrating the dominant nature of this genomic imbalance in cervical cancer. One potential driver gene located on chromosome 3q is the human telomerase RNA component (TERC) gene, which is involved in maintaining the end of chromosomes, the shortening of which is associated with cellular senescence and aging (Heselmeyer-Haddad et al. 2005; Yin et al. 2012). However, one can not overlook that many other genes at chromosome 3q will also exhibit increased transcriptional activity, thus possibly contributing to cervical carcinogenesis. In other tumors of squamous cell origin, such as head and neck or bladder squamous cell carcinoma, the gain of chromosome 3q is also a prevalent alteration. Other genomic imbalances commonly observed in these tumor subtypes include gains located at chromosome 1, 7, and 20, and losses located at 4, 11, 16, 17, and 19 (Cancer Genome Atlas Research Network 2014a).
A catalog of recurrent copy number alterations has also been established for non-small cell lung carcinoma (NSCLC) using aCGH, with specific genomic imbalances including gains at 1q, 3q, 5p, and 8q, and losses at 3p, 8p, 9p, 13, and 17p (Tonon et al. 2005). Although driver genes on genomic sites of copy number changes are still under discovery-based approaches, integrative analysis comprising mutational profiling of NSCLC have identified KRAS, BRAF, EGFR, MET, and FGFR1 as the main candidate driver oncogenes in this disease (Cancer Genome Atlas Research Network 2014b).
Altogether, these studies and others have shown that copy number alterations are tumor-type specific, and that they can be used for efficient tumor classification (Fig. 3). The examples of copy number changes described above suggest that tissue types arising from similar origin tend to share similarities as far as chromosomal gains and losses is concerned, as seen in cancer types of squamous cell origin (i.e., gain of 3q in cervical, bladder, head and neck, NSCLCs), cancers in reproductive organs, such as serous ovarian and serous-like endometrial carcinomas, and cancers affecting gastrointestinal tract tissues (i.e., esophageal, stomach, colon and rectal carcinomas). Comprehensive meta-analyses to understand how tissue or lineage specific transcriptional profiles have an influence on the determination of tumor-type associated signatures of genomic imbalances constitute a fundamental concept underlying the nature of somatic copy number alterations in cancer.
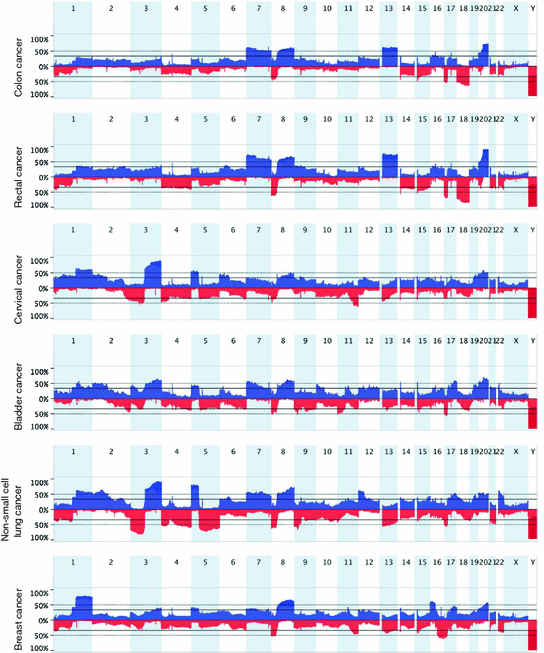
Fig. 3
Diagram of genomic profiles showing the most common gains and losses in colon, rectal, head and neck, bladder and breast cancer. Note the prevalence of specific genomic imbalances unique to each tumor type, thus illustrating the individual landscapes of copy number alterations. SNP array data were collected from The Cancer Genome Atlas (http://cancergenome.nih.gov/)
6 Consequences of Genomic Imbalances on Global Gene Expression
The presence of chromosome imbalances specific to the tissue of cancer origin may lead one to ask: What consequences does this additional genomic material have on the biology of cancer cells? The examples mentioned above suggest that recurrent low-copy number changes provide a selective advantage to the specific cell type to propagate indefinitely, often under sub-optimal metabolic conditions and in the presence of genomic and mitotic defects. In both normal and cancer cells, low-level copy number alterations, regardless if they naturally occur in tumors or are artificially induced in non-tumor cells, result in a massive transcriptional deregulation (Grade et al. 2006b, 2007; Upender et al. 2004) (Fig. 4). In contrast to what has been described in normal cells, aneuploidy-dependent transcriptional enhancement must have a positive impact on the growth of cancer cells (Tang and Amon 2013). In addition, most of the genes implicated in the pathogenesis are located in chromosomal regions selected to give growth advantage to the tumor cell (e.g., gain of MYC at 8q, loss of TP53 at 17p and loss of SMAD4 at 18q in CRC). The integrative strategy of looking at the transcriptional profile for all of the genes confined within regions recurrently involved in genomic imbalances has been extensively used to discover novel cancer genes, as well as to identify genes considered targets for cancer therapeutics (Camps et al. 2013). Therefore, minimal common regions of gains and losses are likely to contain driver genes whose dosage-related deregulation will be of high importance for carcinogenesis.
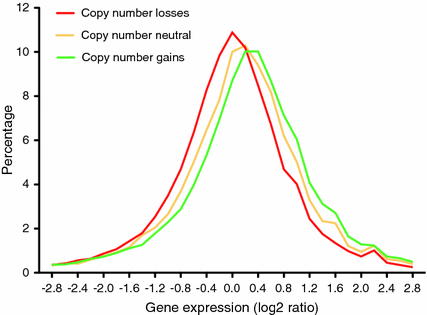
Fig. 4
Plot showing the correlation between copy number changes and gene expression from a set of colorectal cancers. In yellow, genomic segments that are copy number neutral; in red, genomic segments that show a copy number reduction; and in green, genomic segments that show a copy number gain. The Y-axis indicates the levels of gene expression in log2 ratio. (Adapted from Ried et al. 2012)
Nevertheless, it may also be possible that over-activation of proliferative genes, such as the oncogene MYC, impairs cell viability when expressed at “too-high” levels or when affecting the transcription of too many genes, as they induce a disequilibrium of the metabolic stoichiometry of the cancer cell (Sabo et al. 2014; Wahlstrom and Henriksson 2014). Regulation of driver gene expression and their molecular consequences must be critical to ensure cellular viability (Walz et al. 2014). This may be one of the reasons why several cancers do not show high-level amplifications of MYC, but rather show a low-copy number gain of chromosome arm 8q containing this gene to achieve the necessary balance in the amount of this transcription factor (Meyer and Penn 2008). However, one must acknowledge that in such scenarios not only MYC will be over-expressed, but many other genes as well. Thus the question arises as to which extent other genes, e.g., those already described in the literature as drivers, will determine the positive selection of genomic gains (and losses in the case of tumor suppressor genes). For example, the obvious candidate gene for the selection of the 8q gain is MYC, but due to the high recurrence of the low-copy gain of this entire arm, other genes may play a role to boost the cellular fitness. However, whether the transcriptional activation of genes that accompany the target gene of a whole or partial aneuploid chromosome has biological relevance for the cancer cell remains largely uninvestigated. In essence, rather than a single gene, genomic imbalances (i.e., aneuploidy) remarkably represent the driver event in cancer cells.
7 Focal Amplifications and Deletions Point to Driver Genes
7.1 Genomic Amplifications
The term genomic amplification is restricted to focal regions of the genome that are represented in multiple copies (Myllykangas and Knuutila 2006). Two cytogenetically distinct DNA structures have been found to harbor those amplified genomic strings. One type of structure consists of concatenation of a genomic region that has been duplicated numerous times, usually within that same chromosome. These structures fail to display typical banding patterns after trypsin-Giemsa staining and are known as homogeneously staining regions (HSR). The other type of structure consists of small-paired extra-chromosomal bodies known as double minute chromosomes or double minutes (DM) that have been shown to be circular DNA. DMs are basically acentric, atelomeric extra-chromosomal elements containing between 1 and 2 Mb of duplicated DNA that are present in tens to hundreds of copies in a single cell (Kuttler and Mai 2007; L’Abbate et al. 2014). The nature of DMs is still under investigation and it is not clear yet how DMs are inherited from cell to cell. One of the biological mechanisms that originate genomic amplifications is the breakage-fusion-bridge model. Briefly, this model was proposed by Barbara McClintock at the beginning of the last century (McClintock 1939) and is based on the cycling formation of uncapped DNA ends by consecutive DNA double strand breaks and subsequent repair by recombination-based mechanisms, leading to broad DNA amplification, progressive terminal deletions and an increase of genomic instability. These focal regions of high-level copy number change frequently contain oncogenes to promote carcinogenesis (Difilippantonio et al. 2002). The integrative analysis of aCGH and gene expression profiling in cancer allowed the discovery of numerous regions of amplifications in several cancer types, providing evidence for the existence of genes whose oncogenic function was unreported (e.g., Camps et al. 2013; Lockwood et al. 2008). High-throughput analyses of large cohorts of clinical samples resulted in the identification of some cancer lineages with a preference to amplify specific areas of the genome, while other cancer types accumulate low-copy number changes affecting whole chromosomes or chromosome arms (Zack et al. 2013).
Well-known examples of oncogenes that can be activated as a consequence of focal genomic amplification are ERBB2 in breast cancer, MYCN in neuroblastoma, MYC in colon, esophageal, gastric, ovarian cancer and others, CCND1 in bladder cancer, and MDM2 and CDK4 in well-differentiated and dedifferentiated liposarcoma, among others (Crago and Singer 2011). Cyclin D1 (CCND1), located at chromosome band 11q13, plays an important role in cell cycle regulation, binds to cyclin-dependent kinases (CDK4/6), and promotes phosphorylation of RB1, orchestrating progression through the G1 restriction point. This genomic location shows recurrent gene amplifications in several cancer types such as breast, head and neck, bladder, ovarian cancer, and others. Integration of aCGH and gene expression profiling data suggest that CCND1 is not the only target in this recurrent genomic amplification, but that there are other genes within this focal area whose expression might also be relevant for tumorigenesis, suggesting a synergistic effect between well-known driver oncogenes and other genes that are amplified in the same amplicon or in different regions of the genome. For example, PPFIA1 amplification was found exclusively in CCND1-amplified breast cancers, suggesting that PPFIA1 gene copy number changes represent cis-like events of CCND1 amplification (Dancau et al. 2010). Co-amplification in trans at chromosomes 8p11–8p12 and 11q12–11q14, including CCND1, often occurs in breast tumors suggesting a transcriptional crosstalk between genes in the 8p and 11q amplicons, as well as their cooperation with major pathways of tumorigenesis (Kwek et al. 2009). As for probably the most relevant oncogene in human cancer, MYC, the co-amplified neighboring long non-coding RNA gene, PVT1, is essential for maintaining the functional expression of the MYC transcription factor (Huppi et al. 2008; Tseng et al. 2014). Another important oncogenic alteration involves focal amplifications of the FGFR1 gene, located on chromosome 8p and encoding a membrane-bound receptor tyrosine kinase, in up to 20 % of squamous cell lung cancers (Dutt et al. 2011; Weiss et al. 2010).
7.2 Homozygous Deletions
Array CGH played a very important role in the discovery of disease-associated microdeletions with clinical impact, both in developmental-related delays and cancer (Shinawi and Cheung 2008). Perhaps even more important than genomic amplifications in cancer are homozygous deletions, which usually harbor tumor suppressor genes. Tumor suppressor genes are subjected to the two-hit model described by Knudson (1971), where one of the alleles is mutated either in the germline or somatically, while the other allele loses its function either by a second somatic deletion, an epigenetic modification, or by a somatically uniparental disomy event. Among the most frequent losses in human epithelial cancers are the homozygous deletions at 9p21 involving CDKN2A (also referred to as p16), a CDK4 inhibitor, which can also bind the p53-stabilizing protein MDM2 (Ozenne et al. 2010). The absence of functional CDKN2A, either by homozygous deletion, hypermethylation or mutation, contributes significantly to the tumor phenotype through the deregulation of CDK4 and p53, thereby inducing cell cycle G1 progression. CDKN2A is homozygously deleted or hypermethylated at high frequency in cell lines derived from tumors of lung, breast, brain, bone, skin, bladder, kidney, ovary, and lymphocytes (Weisenberger 2014; Gil and Peters 2006).
Analogous to CDKN2A, loss of function for the tumor suppressor gene retinoblastoma 1 (RB1) at 13q14 has similar effects on promoting G1 progression (Manning and Dyson 2011). As previously stated, homozygous deletions at 13q14 have prognostic significance in a variety of not only epithelial human cancers, but also hematological malignancies (Rowntree et al. 2002; Starostik et al. 1999). In colon and rectal cancer, mutations and homozygous deletions of the adenomatous polyposis coli gene (APC) at 5q22, specially in those patients with familial adenomatous syndrome (FAP), but also in a large percentage of sporadic cancer, play a critical role in the release of cytosolic beta-catenin, enabling it to cross the nuclear membrane and transcriptionally activate the Wnt/β-catenin signaling pathway (Nathke 2004), which leads to increased cellular proliferation.
Of note is the high frequency of focal deletions affecting larger genes in the genome, such as FHIT, WWOX, PTPRD, MACROD2, PARK and others, as seen by analyzing somatic copy number alterations in primary tumors in across various cancer cohorts. Since the functional and clinical relevance of these deletions remains elusive, there is no solid evidence that these genes are indeed tumor suppressor genes. Nevertheless, it has been suggested that the genomic plasticity of the regions where these genes are located (e.g., the presence of fragile sites) might be a causative force for these events.
8 Implications for Clinical Practice
As extensively discussed above, chromosomal aberrations play a prominent and defining role in many human solid tumors. Due to the continuous evolution of genomic analysis technologies, cancer genomics has moved from being purely a descriptive enumeration of structural and numerical DNA aberrations, and is increasingly being applied to classify cancer entities into different subtypes (Garraway 2013; Garraway and Lander 2013; McClintock 1939; Tran et al. 2012). For example, screening for amplifications of the MDM2 and CDK4 genes enables classification of well-differentiated and dedifferentiated liposarcomas (Crago and Singer 2011). Furthermore, cancer genomics are employed to identify genetic alterations that can be targeted therapeutically, and therefore may guide clinicians in choosing rational, molecularly defined treatment strategies (Garraway 2013; MacConaill 2013; Tran et al. 2012).
Similar to the standard of care in hematologic malignancies, NSCLC is the prime example of a solid tumor that should undergo extensive molecular biomarker testing prior to starting any treatment (Rampal and Levine 2013). Apart from (activating) mutations in genes such as EGFR, KRAS, BRAF, PIK3CA or DDR2, which do not represent chromosomal aberrations, at least five activating alterations are clinically relevant: ALK, RET, and ROS1 rearrangements, and MET and FGFR1 amplifications (Li et al. 2013; Oxnard et al. 2013; Shames and Wistuba 2014). For ALK, which encodes a transmembrane tyrosine-kinase receptor, oncogenic fusion with one of its upstream activators, EML4, has been demonstrated (Soda et al. 2007). While advanced tumors with EML4–ALK rearrangements are insensitive to EGFR tyrosine-kinase inhibitors (TKIs) often used for treating NSCLC (Shaw et al. 2009), high initial responses have been observed upon treatment with the ALK inhibitor crizotinib (Kwak et al. 2010; Shaw et al. 2013). Nevertheless, acquired resistance to crizotinib frequently develops (Shaw and Engelman 2013), and evidence is accumulating that this secondary resistance can be overcome with novel ALK inhibitors such as ceritinib (Shaw and Engelman 2014). Several different fusion partners of the gene encoding the tyrosine-kinase receptor ROS1 have been identified (Bergethon et al. 2012; Rimkunas et al. 2012; Takeuchi et al. 2012), and ROS1-rearranged tumors may also benefit from treatment with crizotinib (Bos et al. 2013).
Similarly, fusions between the tyrosine-kinase receptor gene RET have been reported with KIF5B, CCDC6 or TRIM33 (Ju et al. 2012; Kohno et al. 2012; Lipson et al. 2012; Takeuchi et al. 2012). Because several TKIs show at least some activity against the RET kinase, RET-rearranged tumors are amenable to targeted strategies as well (Oxnard et al. 2013). Another prominent alteration involves amplification of the membrane-bound tyrosine receptor gene FGFR1, which led to the development of FGFR TKIs (Dutt et al. 2011; Gavine et al. 2012; Weiss et al. 2010; Zhang et al. 2012). Interestingly, MET amplifications have been associated with acquired resistance of EGFR-mutated NSCLC to TKIs (Cappuzzo et al. 2009; Dziadziuszko et al. 2012; Toschi and Cappuzzo 2010). Because amplifications in the MET gene activate multiple signaling pathways, a number of agents targeting this transmembrane tyrosine-kinase receptor are in preclinical and clinical development (Sadiq and Salgia 2013). Unfortunately, however, it was quickly realized that targeted agents developed for adenocarcinoma, the most common type of lung cancer, were largely ineffective against squamous cell carcinoma, the second most common type of lung cancer.
Related posts:
 Modelling: What Have Mouse Models for Chromosome Instability Taught Us?
Modelling: What Have Mouse Models for Chromosome Instability Taught Us?
 of Chromosomal Instability
of Chromosomal Instability
 of Aneuploidy in Cancer: Transcriptome and Beyond
of Aneuploidy in Cancer: Transcriptome and Beyond
 Diverse Effects of Complex Chromosome Rearrangements and Chromothripsis in Cancer Development
Diverse Effects of Complex Chromosome Rearrangements and Chromothripsis in Cancer Development
 Dysfunction, Chromosomal Instability and Cancer
Dysfunction, Chromosomal Instability and Cancer
 as Models of Mitotic Fidelity
as Models of Mitotic Fidelity
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


