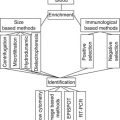
Fig. 6.1
Bioactive food components and its integration with various molecular streams in phenotype progression and development
Table 6.1
Factors affecting nutrigenomic research and phenotype
Approach | Definition and factors |
|---|---|
Transcriptomics | The branch of molecular biology that deals with the study of messenger RNA molecules produced in an individual or population of a particular cell type |
Identification of transcription factors that respond to nutrients and gene targets | |
RNA amplification and procedure (quantity, quality, replicates, real-time PCR, high-density analysis) | |
Quantity of starting tissue/cell material | |
Fold change in expression | |
Intraindividual and interindividual variations in healthy and diseased subjects and Identification of single genes or group of genes that are regulated (up or down) in a particular disease or nutritional condition | |
Heterogeneity of cell populations and single cell gene expression profiling | |
Combination of gene variants (SNPs) | |
Data processing and interpretation | |
Epigenetics | It is defined as heritable changes in gene expression, which are not due to any alteration in the DNA sequence |
Characterization of chromatin modifications that influence gene expression and impact of nutrients | |
Histone modifications | |
DNA methylation | |
Nucleosome organization | |
Order, interdependence and intradependence, and reversibility of histone modifications | |
Cross talk and mutual dependency between histone modifications, DNA methylation, and methyl-binding proteins | |
Help in exploring the evolutionary origin of cell differentiation and change in cancer cells | |
Proteomics | It is the large-scale study of proteins, particularly their structures and functions |
Linking gene expression studies with protein functions | |
Tissue and cellular localization | |
Plasma levels | |
Expression levels | |
Posttranslational modifications | |
Protein–protein interactions | |
Cellular function | |
Bioinformatics and data interpretations | |
Metabolomics | It is the scientific study of chemical processes involving metabolites |
Linking exposure to biological effects induced by metabolites | |
Interindividual differences in metabolisms and disposition | |
Measurements of metabolite in specimens | |
Recovery methods from tissue/plasma |
Exploitation of this genomic information, along with high-throughput “omic” technologies, allow the acquisition of new knowledge aimed at obtaining a better understanding of nutrient–gene interactions depending on the genotype, with the ultimate goal of developing personalized nutrition strategies for optimal health and disease prevention [8]. There are three central factors that underpin nutrigenetics and nutrigenomics as a central science. First there is great diversity in the inherited genome between ethnic groups and individuals which affects nutrient bioavailability and metabolism. Second, people differ greatly in their food/nutrient availability and choices depending on cultural, economic, geographical, and taste perception differences. Third, malnutrition (deficiency or excess) itself can influence gene expression and genome stability; the latter leading to mutations at the gene sequence or chromosomal level, which may cause abnormal gene dosage and gene expression leading to adverse phenotypes during the various life stages [9].
The decisive goal is to (1) match the nutriome (i.e., nutrient intake combination) with the current genome status (i.e., inherited and acquired genome) so that genome maintenance, gene expression, metabolism, and cell function can occur normally and in a homeostatically sustainable manner [8] and (2) provide better interpretation of data from epidemiological and clinical intervention studies regarding health impacts of dietary factors that may help to revise recommendations for personalized nutrition [10].
The fundamental hypotheses reinforcing the science of nutrigenetics and nutrigenomics are the following [9]:
Nutrition may apply its impact on health outcomes by directly affecting expression of genes in critical metabolic pathways and/or indirectly by affecting the incidence of genetic mutation at the base sequence or chromosomal level which in turn causes alterations in gene dosage and gene expression.
The health effects of nutrients and nutriomes (nutrient combinations) depend on inherited genetic variants that alter the uptake and metabolism of nutrients and/or the molecular interaction of enzymes with their nutrient cofactor and hence the activity of biochemical reactions.
Better health outcomes can be achieved if nutritional requirements are customized for each individual, taking into consideration both his/her inherited and acquired genetic characteristics depending on life stage, dietary preferences, and health status.
Genomic and epigenomic processes likely do not utterly account for the ability of dietary factors to influence phenotypic changes, since changes in the rate of transcription of genes (transcriptomics) can also be fundamental to cellular processes [11]. Multiple pathways appear to overlap as a cause of multiple diseases [12]. Thus, the examination of these pathways via transcriptomic profiles may simultaneously provide important hints about multiple disease risks. Noteworthy, several bioactive food components, including both essential and nonessential nutrients, can control gene expression patterns. Their influence on gene transcription and translation is not only concentration dependent but also time dependent [13]. Yet these changes may provide significant insights about the specificity of individual food components to influence one or more biological processes, including those involved in the risk of cancer development and/or tumor behavior.
This chapter will try to clarify the current research updates on interaction of diet with genetic backgrounds and how diet is involved in the development of one of the most common cancers afflicting females today globally. Simultaneously we will pinpoint the various diets that can be used in curing or decreasing the risks, although the nature of these interactions is indeed very complex.
Nutrigenomic Diseases
Diseases that are known to be associated with the interactions between multiple genetic and environmental factors such as diet include many cancers, diabetes, heart disease, obesity, and some psychiatric disorders. Therefore, both disciplines aim to unravel diet–genome interactions; however, their approaches and immediate objectives are distinct. Nutrigenomics will unravel the optimal diet from within a series of nutritional alternatives, whereas nutrigenetics will yield critically important information that will assist clinicians in identifying the optimal diet for a given individual, i.e., personalized nutrition [10]. The following five tenets of nutritional genomics serve as a conceptual basis for understanding the focus and promise of this budding field: [7]
1.
Under certain conditions and in some individuals, diet can be a serious risk factor for a number of diseases.
2.
Universal dietary chemicals can act on the human genome, either directly or indirectly, to alter gene expression or structure.
3.
The degree to which diet influences the balance between healthy and disease states may depend on a person’s genetic makeup.
4.
Some diet-modulated genes (and their normal, common variants) probably play a role in the onset, incidence, progression, and/or severity of chronic diseases.
5.
Dietary intervention based on the knowledge of nutritional requirements, nutritional status, and genotype (i.e., personalized nutrition) can be employed to prevent, mitigate, or cure chronic disease.
Nutrigenomics and Carcinogenesis
Cancer is a process composed of multiple stages in which gene expression, and protein and metabolite function, begins to operate aberrantly [14]. In the post-genomic era, the cellular events mediating the onset of carcinogenesis, in addition to their modulation by dietary factors, have yielded significant information in understanding of this disease [15]. Inherited mutations in genes can increase one’s susceptibility for cancer. Evidences of genome and epigenome damage biomarkers, in the absence of overt exposure of genotoxins, are themselves sensitive indicators of deficiency in micronutrients required as cofactors or as components of DNA repair enzymes, for maintenance methylation of CpG sequences and prevention of DNA oxidation and/or uracil incorporation into DNA [16]. Diet is considered as a source of either carcinogens (intrinsic or cooking generated) present in certain foods or constituents acting in a protective manner (vitamins, antioxidants, detoxifying enzyme-activating substances, etc.) [17]. It is clear that carcinogen metabolism-affecting polymorphisms may modify the chance of contact between carcinogens and target cells, thus acting at the stage of cancer initiation. Influences of polymorphisms of gene encoding factors involved in hormonal regulation are most strongly manifested in hormone-dependent tumors such as breast, prostate, ovarian, and endometrial cancers. Polymorphisms in sex hormone receptor genes comprising those encoding estrogen receptor, progesterone receptor, and androgen receptor have been shown to be associated with cancer risk modulation [18]. Dietary factors can undoubtedly interact with hormonal regulation. Obesity strongly affects hormonal status. At the same time, some food components, such as phytoestrogens, are known to be processed by the same metabolic pathways as sex hormones [19]; thus their cancer-preventive effect can be modulated by the polymorphisms.
Epigenetic Link with Nutrigenomics
An important emerging part of nutrient–gene interaction studies with the potential for both intra- and transgenerational effects is epigenetics [20]. Epigenetics refers to the processes that regulate how and when certain genes are turned on and off, while epigenomics pertains to analysis of epigenetic changes in a cell or the entire organism. Epigenetic processes have a sturdy influence on normal growth and development, and this process is deregulated in diseases such as cancer. Diet on its own or by interaction with other environmental factors can cause epigenetic changes that may turn certain genes on or off. Table 6.2 provides a brief description of the nutrients and chemicals involved in DNA methylation. Epigenetic silencing of genes that would usually protect against a disease, as a result, could make people more susceptible to developing that disease later in life. The epigenome, which is heritable and modifiable by diet, is the global epigenetic pattern determined by global and gene-specific DNA methylation, histone modifications, and chromatin-associated proteins that control expression of housekeeping genes and restrains the expression of parasitic DNA such as transposons. Table 6.3 represents the dietary chemicals, DNA methylation, and its mechanism of action.
Table 6.2
Nutrients and chemicals involved in DNA (hyper-/hypo-) methylation
Nutrient | Chemicals |
|---|---|
Alcohol | Genistein |
Arsenic | Methionine |
Betaine | Nickel |
Cadmium | Polyphenol |
Choline | Selenium |
Coumestrol | Vitamin A |
Equol | Vitamin B6 |
Fiber | Vitamin B12 |
Folate | Zinc |
Table 6.3
Dietary chemicals, DNA methylation, and its mechanism of action
Dietary chemicals | Mechanism of action | Phenotype/outcome | Reference |
|---|---|---|---|
Alcohol | Affects folate metabolism, altering DNA methylation | Cancer susceptibility | [23] |
Arsenic | Compete with cytosine, DNA methyl transferase and selenium for methyl donation from S-adenosil-1-methionine | Global hypomethylation in the liver, cancer susceptibility | [24] |
Choline | Deficiency in diets has been associated with decreased tissue S-adenosil-1-methionine | Hepatic tumorigenesis, cancer susceptibility | [25] |
Folate | Its deficiency has complex effect on DNA methylation depending on cell type, organ, and development stage | Cancer susceptibility | [26] |
Genistein | Dietary genistein can migrate tumorigenic process via promoter modulation of gene expression | Mitigates tumorigenesis | [27] |
Lycopene | It has direct DNA demethylating activity. It migrates tumorigenic processes via promoter methylation modulation of gene expression | Mitigates tumorigenesis | [28] |
Methionine | Its deficiency decreases tissue SAM resulting in global DNA hypomethylation and HCC in rodents | HCC | [29] |
Nickel | Environmental carcinogen, induce de novo methylation of tumor-suppressor genes Suppressive effect on histone H4 acetylation in mammalian cells | Cancer susceptibility | [30] |
Selenium | Its deficiency decreases DNA methylation. Low intake influences the activity of selenoproteins, causing changes in mRNA levels for the encoding genes | Cancer susceptibility | [31] |
Vitamins | Vitamins (B2, B6, and B12) are necessary cofactors in one carbon (methyl metabolism) | Affect several metabolic pathways, cancer susceptibility | [32] |
One study has demonstrated that sulforaphane, butyrate, and allyl sulfur are effective inhibitors of histone deacetylase (HDAC). HDAC inhibition was associated with global increases in histone acetylation, enhanced interactions of acetylated histones with the promoter regions of the P21 and BAX genes, and higher expression of p21Cip1/Waf1 and BAX proteins [33]. Importantly, sulforaphane has been reported to reduce HDAC activity in humans [33]. Future research likely needs to relate HDAC changes in humans to a change in a cancer-related process. Furthermore, since acetylation is only one method to regulate histone homeostasis [34], greater concentration needs to be given to how nutrition might influence the other types of histone modifications.
The field of nutrigenomics harnesses multiple disciplines and includes dietary effects on genome stability (DNA damage at the molecular and chromosome level), epigenome alterations (DNA methylation), RNA and micro-RNA expression (transcriptomics), protein expression (proteomics), and metabolite changes (metabolomics), all of which can be studied independently or in an integrated manner to diagnose health status and/or disease trajectory. However, of these biomarkers, only DNA damage is a clear biomarker of fundamental pathology that may be mitigated by promotion of apoptosis of genetically aberrant cells or by reducing the rate of DNA damage accumulation. Changes at the epigenome, transcriptome, proteome, and metabolome levels may simply reflect modifiable homeostatic responses to altered nutritional exposure and on their own may not be sufficient to indicate definite irreversible pathology at the genome level.
DNA damage can be diagnosed in a number of complementary ways as follows: (1) damage to single bases (e.g., DNA adducts such as the addition of a hydroxyl radical to guanine caused by oxidative stress), (2) basic sites in the DNA sequence (measurable by use of the aldehyde-reactive probe), (3) DNA strand breaks (commonly measured using the Comet assay), (4) telomere shortening (measured by terminal restriction fragment length analysis, quantitative PCR, or flow cytometry), (5) chromosome breakage or loss (usually measured using micronucleus cytome assays or metaphase chromosome analysis), and (6) mitochondrial DNA damage (usually measured as deletions or base damage in the circular mitochondrial DNA sequence). These DNA damage biomarkers are presently at different levels of validation based on evidence relating to the association with nutrition (cross-sectional epidemiological and intervention studies) and disease (cross-sectional epidemiology and prospective cohort studies) [35]. The micronucleus assay in cytokinesis-blocked lymphocytes is currently the best validated biomarker for nutritional genomic studies of DNA damage.
Given the advances in diagnostic technologies assessing DNA damage, it has now become feasible to determine dietary reference values for DNA damage prevention and to start translating into practice the Genome Health Clinic concept of DNA damage prevention [35]. The latter is based on the recognition that damage to the genome is the most elementary cause of developmental and degenerative diseases, which can be accurately diagnosed and prevented by appropriate diet and lifestyle intervention at a genetic subgroup and personalized level. The ability of diet to affect the flow of genetic information can occur at multiple sites of regulation [10]. Advances in genomics, transcriptomics, proteomics, and metabolomics have enabled a more rapid and comprehensive understanding of how bioactive compounds affect human health. Dietary bioactive compounds can be tested for their potential health-promoting properties by applying these different technologies to cell culture, and animal or human studies. Each experimental approach offers unique strengths and has certain limitations.
Current Updates of Nutrigenomic Studies in Breast Cancer
Several studies of sporadic breast cancers have shown that fruits and vegetables [36], fish, monounsaturated and polyunsaturated fatty acids [37], vitamin D, calcium, and phytoestrogens may reduce the risk of breast cancer, although there are inconsistencies in the literature. High intake of meat, poultry, total energy, and total fat and saturated fatty acids has been reported to be associated with increased risk for breast cancer [38]. Malmö Diet and Cancer cohort examined the association between dietary folate equivalents (DFE) and breast cancer among carriers of two genetic polymorphisms for MTHFR gene (MTHFR 677C/T and 1298A/C). A positive association between DFE and breast cancer among women carriers of MTHFR 677CT/TT-1298AA occurred while an inverse association was observed in 677CT-1298 AC women [39]. In a nested case-control study, Maruti et al. reported that postmenopausal women with two copies of variant T alleles (TT genotype) had increased risk of breast cancer. In addition, the intake of other B vitamins may influence the relationship between the MTHFR genetic variants and breast cancer risk. It has been found that the most pronounced MTHFR-breast cancer risk was observed among women with the lowest intakes of dietary folate and vitamin B6 [40].
In a nested case-control study within the Singapore Chinese Health Study, it has been observed that there is an inverse relationship between breast cancer risk and low folate intake and weekly/daily green tea intake compared with reduced green tea intake. Also, women carrying the high-activity MTHFR/TYMS genotypes with 0–1 variant allele and weekly/daily green tea intake had a lower breast cancer risk, especially women who also had low folate intake. No association was observed among women carriers of two variant alleles. These findings suggest one of the mechanisms through which green tea can provide protection against breast cancer is through folate regulation [41]. The anticancer effect of the EGCG (tea polyphenol (−)-epigallocatechin-3-gallate) may be mediated by the regulation of epigenetic processes. It was found that EGCG can lead to ERα reactivation in the ERα-negative breast cancer cell lines by remodeling the chromatin structure of the ERα promoter by the alteration of the histone acetylation and methylation status [42]. Further, it has been reported that EGCG inhibits the telomerase by decreasing the hTERT promoter methylation and ablating the histone H3 Lys9 acetylation in the MCF-7 cell lines [43].
Another study reported the inverse relationship between green tea intake and breast cancer risk among Asian-Americans [44] as a function of the catechol-O-methyltransferase (COMT) genotype. This enzyme is recognized to be involved in the metabolism of the tea polyphenols. More specifically, a reduced risk of breast cancer was observed only among tea (green and black) drinker carriers of at least one low-activity COMT allele. These findings of reduced breast cancer risk with tea catechins, especially in women who had the low-activity COMT alleles, suggest that these women were less efficient in eliminating tea catechins, therefore optimizing the benefits from the tea and its associated bioactive constituent [45].
The Shanghai Breast Cancer Study examined the relationship between breast cancer risk, GSTP1 genetic variants, and other diet components, the cruciferous vegetables. The GSTP1 Val/Val genotype was associated with increased breast cancer risk, especially in premenopausal women with low intake of cruciferous vegetables. Thus, cruciferous vegetable intake with high isothiocyanates may reduce breast cancer risk and modify the effect of the GSTP1 genotype [45], and not necessarily be beneficial to all women.
Finally, the response to marine n-3 fatty acids with breast cancer risk may depend on genetics. Women with genetic variants that encode lower or no enzymatic activity of GSTT1 have a 30 % lower breast cancer risk from the marine n-3 fatty acids, when compared with women with high-activity genotypes. These data suggest that the peroxidation products of n-3 fatty acids may be involved in the protection against breast cancer [46]. This is not that unusual since other food components have been reported to inhibit tumors by generating free radicals [47]. Watercress, a rich source of phenethyl isothiocyanates (PEITC), has been proposed to have anticancer activity. A crude watercress extract was reported to inhibit cancer cell growth and hypoxia-inducible factor (HIF) activity and reduced angiogenesis by decreasing the phosphorylation of the translation regulator 4E binding protein 1 (4E-BP1) [48].
In a breast case-control study that examined the association of the 5-lipoxygenase gene (ALOX) and 5-lipoxygenase-activating protein gene (ALOX5AP) polymorphisms and dietary linoleic acid intake with breast cancer risk, it was found that women carriers of two variant alleles for the ALOX5AP 4900 A/G who had a diet rich in linoleic acid had a greater breast cancer risk compared with women carrying AG or GG genotypes. These results propose that genetic predisposition related to n-6 polyunsaturated fatty acid metabolism should be taken into account when the relation between dietary fat and breast cancer risk is examined [49]. Furthermore, dietary omega-3 polyunsaturated fatty acids downregulate the expression of the polycomb group (PcG) protein, enhancer of zeste homologue 2 (EZH2) in breast cancer cells. This study reported a decrease in histone 3 lysine 27 trimethylation (H3K27me3) activity of EZH2 and upregulation of E-cadherin and insulin-like growth factor-binding protein 3. The treatment with omega-3 PUFAs led to decrease in the invasion capacity of the breast cancer cells [50].
Additionally, a nested case-control study of postmenopausal women examined the interaction between oxidative stress-related genes including catalase (CAT) C262T, myeloperoxidase (MPO) G463A, endothelial nitric oxide synthase (NOS3) G894T, and heme oxygenase-1 (HO-1) GT (n) dinucleotide length polymorphism and level of vegetable and fruit intake on breast cancer risk. The study found that women with low intake of vegetables and fruits and the low-risk CAT CC genotypes appeared to be associated with increased breast cancer risk, especially those women with four or more low-risk alleles, suggestive of the role of endogenous and exogenous antioxidants in breast carcinogenesis [51].
Iwasaki et al. examined the effect of four SNPs in cytochrome P450c17alpha (CYP17), aromatase (CYP19), 17beta-hydroxysteroid dehydrogenase type I (17beta-HSD1), and sex hormone-binding globulin (SHBG) genes on the association between isoflavone intake and breast cancer risk. The study identified an inverse association between isoflavone intake and breast cancer risk among women with at least one variant allele for the 17beta-HSD1 polymorphism and among postmenopausal Japanese women with GG genotype for the SHBG gene, indicating that genetic variants of the 17beta-HSD1 and SHBG genes may modify the relationship between isoflavone intake and breast cancer risk [52]. The association between isoflavones and breast cancer risk may be explained by the antiestrogenic effect of the isoflavones and the effect on the DNA methylation. It has been reported that the daily administration of isoflavones to healthy premenopausal women led to dose-specific changes in RARbeta2 and CCND2 gene promoter methylation, changes that correlated with genistein levels. Additionally, an inverse correlation between estrogenic marker complement C3 and genistein was observed, suggesting an antiestrogenic effect [53]. Furthermore, genistein has been associated with specific epigenetic changes. For example, the long-term exposure to genistein of the MCF-7 breast cancer cell lines has been found to lead to reduced expression of the acetylated histone 3 (H3). Additionally, this exposure was associated with alteration in growth responses to mitogenic factors and histone deacetylase inhibitors [54].
Dietary berries have been suggested to influence breast cancer risk (AICR Report), although considerable variability in response is observed. Preclinical studies demonstrate that tumor formation is suppressed as a result of the levels of E(2)-metabolizing enzymes during the early phase of E(2) carcinogenesis [55].
A nested case-control study within the Singapore Chinese Health Study Cohort showed a significant interaction between the level of green tea drinking and the activity of the angiotensin-converting enzyme (ACE) with respect to breast cancer risk depending on ACE gene polymorphism [56]. Even the BRCA breast cancer-associated gene is affected by diet. A diet rich in fruits and vegetables protects a woman from the BRCA gene becoming activated [57]. Olive oil is an integral ingredient of the “Mediterranean diet” and accumulating evidence suggests that it may have a potential role in lowering the risk of several types of cancers. A number of epidemiological studies have linked consumption of olive oil with a reduced risk of cancer, and researchers are increasingly investigating this association further in laboratory studies. The mechanisms by which the cancer-preventing effects of olive oil as having novel anticancer actions may relate to the ability of its monounsaturated fatty acid (MUFA) oleic acid (OA; 18:1n-9) to specifically regulate cancer-related oncogenes. Exogenous supplementation of cultured breast cancer cells with physiological concentrations of OA was found to suppress the overexpression of HER2 (Her-2/neu, erbB-2), a well-characterized oncogene playing a key role in the etiology, progression, and response to chemotherapy and endocrine therapy in approximately 20 % of breast carcinomas.
OA treatment was also found to synergistically enhance the efficacy of trastuzumab (Herceptin) a humanized monoclonal antibody binding with high affinity to the ectodomain (ECD) of the Her2-coded p185(HER2) oncoprotein. Moreover, OA exposure significantly diminished the proteolytic cleavage of the ECD of HER2 and, consequently, its activation status, a crucial molecular event that determines both the aggressive behavior and the response to trastuzumab of Her2-overexpressing breast carcinomas. Recent findings further reveal that OA exposure may suppresses HER2 at the transcriptional level by upregulating the expression of the Ets protein PEA3 -a DNA-binding protein that specifically blocks HER2 promoter activity in breast, ovarian, and stomach cancer cell lines. This anti-HER2 property of OA offers a previously unrecognized molecular mechanism by which olive oil may regulate the malignant behavior of cancer cells. From a clinical perspective, it could provide an effective means of influencing the outcome of Her-2/neu-overexpressing human carcinomas with poor prognosis. Indeed, OA-induced transcriptional repression of HER2 oncogene may represent a novel genomic explanation linking olive oil and cancer, as it seems to equally operate in various types of Her-2/neu-related carcinomas [58].
In another study, OA treatment in Her-2/neu-overexpressing cancer cells was found to induce upregulation of the Ets protein polyomavirus enhancer activator 3 (PEA3), a transcriptional repressor of Her-2/neu promoter. Also, an intact PEA3 DNA-binding site at endogenous Her-2/neu gene promoter was essential for OA-induced repression of this gene. Moreover, OA treatment failed to decrease Her-2/neu protein levels in MCF-7/Her2-18 transfectants, which stably express full-length human Her-2/neu cDNA controlled by a SV40 viral promoter. OA-induced transcriptional repression of Her-2/neu occurs through the action of PEA3 protein at the promoter level [59].
Various Diet Components and Their Cellular/Molecular Effects on Breast Cancer
All main signaling pathways are deregulated in cancer, including cell proliferation, apoptosis, DNA repair, carcinogen metabolism, inflammation, immunity, differentiation, and angiogenesis. Increasingly, evidence points to each of these as molecular targets for cancer prevention. Since several of these sites appear to be tailored by multiple dietary components, it becomes challenging to tease apart nutrient–nutrient interactions and thus what constitutes an ideal diet for health promotion [2]. For example, the apoptosis or programmed cell death that is essential in the fight against cancer through two pathways—either the intrinsic, mitochondrial-mediated pathway, or the extrinsic, death receptor-mediated pathway—could be a target for dietary bioactive agents including genistein, curcumin, resveratrol, luteolin, lupeol, indole 3-carbinol, etc. [4]. Dietary components can modulate apoptosis through effects at different levels, all of which culminate in changes in gene expression. Table 6.4 provides details on transcription factor pathways mediating nutrient–gene interactions.
Macronutrients | Compound | Transcription factor |
|---|---|---|
Fats | Fatty acids | PPARs, SREBPs, LXR, HNF4, ChREBPs, LRs, FXR |
Cholesterol | ||
Carbohydrates | Glucose | USFs, SREBPs, ChREBP |
Proteins | Amino acids | C/EBPs |
Micronutrients | ||
Vitamins | Vitamin A | RAR, RXR, VDR, PXR |
Vitamin D | ||
Vitamin E | ||
Minerals | Calcium | Calcineurin/NF-ATs |
Iron | IRO1, IRP2 | |
Zinc | MTF-1 | |
Other food components | ||
Soy | Flavonoids | ER, NF-Kb, AP1 |
Xenobiotics | CAR, PXR | |
Compounds such as Japanese knotweed (Polygonum c.), 20 % resveratrol, ginger, (Zingiber off.) 5 % gingerols, Rosemary (Rosemarinus off.), 6 % carnosic acid, 1 % rosemarinic acid, 1.5 % ursolic aicd, etc have demonstrated broad-spectrum, multi-targeting, anticancer effects, as well as disease-preventive and health-promoting benefits. The other important aspect of these compound-rich foods, spices, and herbs is that have been regularly used by many cultures throughout the world. Cancer prevention studies have exposed that all of the major signaling pathways deregulated in different types of cancer are affected by nutrients. Pathways studied include carcinogen metabolism, DNA repair, cell proliferation/apoptosis, differentiation, inflammation, oxidant/antioxidant balance, and angiogenesis [61].
So far, more than 1,000 different phytochemicals have been identified with cancer-preventive activities [62]. Dietary fibers have a protective effect against bowel cancer. Long-chain polyunsaturated fatty acids (LC-PUFA) beneficially affect physiological processes, including growth; neurological development; lean and fat mass accretion; reproduction; innate and acquired immunity; infectious pathologies of viruses, bacteria, and parasites; and the incidence and severity of virtually all chronic and degenerative diseases including cancer, atherosclerosis, stroke, arthritis, diabetes, osteoporosis, neurodegenerative, inflammatory, and skin diseases [63]. Fish oil, rich in omega-3 fatty acids, inhibits the growth of colonic tumors in both in vitro and in vivo systems [64].
Bioactive components present in fruits and vegetables can prevent carcinogenesis by several mechanisms, such as blocking metabolic activation through increasing detoxification. Plant foods can modulate detoxification enzymes as flavonoids, phenols, isothiocyanates, allyl sulfur compounds, indoles, and selenium [65]. As a result of carcinogen activation, covalent adducts with the individual nucleic acids of DNA or RNA are formed. It has also been found that reactive oxygen species (ROS) such as superoxide anions, hydrogen peroxide, and hydroxyl radicals attack DNA bases, resulting in potential mistranscription of DNA sequence [66]. Such disruptions can interfere with DNA replication and thus produce mutations in oncogenes and tumor-suppressor genes. ROS can also result in breakage of DNA strand, resulting in mutations or deletions of genetic material [67].
Vitamin D
Few findings demonstrate that primary circulating form of vitamin D 1,25(OH)2D acts in a cell type and tissue-specific manner. For example, 1,25(OH)2D inhibits cell growth of both normal and tumor cells by inhibiting the transition for the G1 to the S phase of the cell cycle [68]. This effect was mediated by increased expression of cyclin A1 in ovarian cancer cells [69], whereas breast cancer cells had increased expression of cyclin D2 [70]. It has also been suggested that the enzyme responsible for the degradation of vitamin D metabolites, CYP24, can also be influenced by cancer. The CYP24 gene was amplified in breast tumors [71].
The majorities of established breast cancer cell lines express transcriptionally active VDR and undergo growth inhibition in response to 1,25D [72]. In general, VDR expression and sensitivity to 1,25D-mediated growth arrest is higher in the less aggressive, estrogen receptor (ER)-positive breast cancer cell lines such as MCF-7 than in ER-negative cell lines. Tumor cells derived from VDR null mice were used to conclusively demonstrate that 1,25D mediates effects in breast cancer cells via the nuclear VDR [73]. Screening for molecular changes induced by 1,25D or vitamin D analogs in various breast cancer cells has identified scores of VDR regulated genes and proteins in diverse pathways, indicating a broad range of downstream [74] involved in cell cycle (cyclins, cyclin-dependent kinases and their inhibitors), apoptosis/autophagy (bcl-2 family, caspases, cathepsins), and inflammation (NFkB, prostaglandins, cox-2). The net effect of these changes is to block mitogenic signaling, including that of estrogen, EGF, IGF-1, and KGF, and to enhance the effects of negative growth factors such as TGFb. In many breast cancer cell lines, 1,25D-mediated growth arrest is associated with the induction of differentiation markers such as casein, lipid droplets, and adhesion proteins [75]. Notably, 1,25D exerts additive or synergistic effects in combination with other triggers of apoptosis, such as ionizing radiation and chemotherapeutic agents [76].
Collectively, these studies indicate that a wide variety of signaling pathways, cell cycle and apoptotic regulatory proteins, and proteases contribute to the antiproliferative, pro-differentiating, and apoptotic effects of 1,25D depending on the specific breast cancer cell line and/or context. In primary cultures of normal human mammary epithelial (HME) cells, vitamin D signaling also mediates growth arrest and induction of differentiation markers such as E-cadherin, but apoptosis has not been observed [77]. In contrast to breast cancer cells, non-transformed mammary cells retain expression of CYP27B1 and generate 1,25D when incubated with physiological concentrations of 25D. Many breast cells also express the megalin–cubilin complex, which mediates internalization of 25D bound to the vitamin D binding protein [78]. Autocrine metabolism of 25D triggers chemopreventive effects in breast epithelial cells including growth inhibition, differentiation, and protection from various cellular stresses [77].
In the intact mammary gland, the epithelium is surrounded by stromal fibroblasts and adipocytes, which provide critical growth factor signals for development and also impact on carcinogenesis. Recent evidence suggests that breast adipocytes express CYP27B1 and generate 25D, which signals via adipocyte VDR to release inhibitory factors that regulate mammary epithelial cell growth [79]. Since vitamin D metabolites are stored in fat tissue, the contribution of adipocyte signaling to the tumor-suppressive actions of vitamin D in mammary gland are likely of physiological importance and require further study. As in colon cancer, acquisition of the transformed phenotype in breast cells is associated with deregulation of the vitamin D pathway [80]. In HME cells, introduction of SV40 large T antigen and/or oncogenic ras induces transformation and reduces responsiveness to 25D in association with downregulation of VDR and CYP27B1 [81]. Oncogenes and tumor-suppressor genes that impact on VDR expression in breast cells include ras, p53, and slug, which act via diverse mechanisms including transcriptional regulation and mRNA instability [82]. The mechanism by which transformation abrogates CYP27B1 expression in breast cells is not yet known.
Green Tea
From a nutrigenomic perspective, the use of green tea as a nutraceutical or functional food has shown anticancer potential, in particular for breast cancer, although more studies are needed. High levels of angiotensin II have been associated with an increased risk of breast cancer development in humans [56]. The angiotensin I-converting enzyme (ACE) gene encodes or activates the enzyme that converts angiotensin I to the active angiotensin II. A low conversion rate is linked with lower rates of breast cancer in women than in those who have a high conversion rate. In those with the high-activity genotype, a high consumption of green tea has resulted in a dramatic drop of one-third in the risk of developing breast cancer. The authors concluded that the antioxidant properties (particularly of the EGCG) are protective against the reactive oxygen species (or free radicals) generated by the high levels of angiotensin II. No such association was made in women with the low levels of angiotensin. Epigallocatechin gallate (ECGC) and other green and black tea polyphenols inhibit cancer cell survival. EGCG suppressed androgen receptor expression and signaling via several growth factor receptors. Cell cycle arrest or apoptosis involved caspase activation and altered Bcl-2 family member expression. EGCG inhibited telomerase activity and led to telomere fragmentation. While at high concentrations polyphenols had pro-oxidative activities, at much lower levels, antioxidative effects occurred [83].
Another study conducted at the University of Southern California looked at green tea ingestion and activity of the catechol-O methyl transferase (COMT) gene [45]. Dr Wu’s group also found an association between green tea intake and a cancer-protective effect in those individuals with at least one copy of the low conversion COMT gene. This means that the beneficial catechins remained in circulation for a longer time period and reduced the risk of breast cancer. It is important to note that the study was only conducted in Asian-Americans and needs to be reproduced in a wider population group.
Soy and Isoflavones
The interest on isoflavones in breast cancer prevention derives from the fact that breast cancer risk for women residing in geographical areas of high consumption of soy products during puberty is lower compared to that of women living in Western countries, and Asian women who had a low soy intake [84]. Table 6.5 gives details on various studies on isoflavones and nutrigenomic approaches in breast cancers. However, clinical trials reported small [90] or no effect of supplementation with isoflavones on breast cancer risk [91], and administration of isoflavones elicited in some cases an estrogen-like effect. Other studies indicated that the reduction in breast cancer risk due to soy intake was limited to Asian populations [92]. A case-control study conducted in Southeast China in 2004–2005 reported that premenopausal and postmenopausal women in the highest quartile of total isoflavone intake had a reduced risk for all receptor (ER/PR) status of breast cancer with a dose–response relationship. The protective effect was more pronounced for women with ER+/PR + and ER–/PR– breast tumors [93].
Table 6.5
Various studies on isoflavones and nutrigenomic approaches in breast cancers