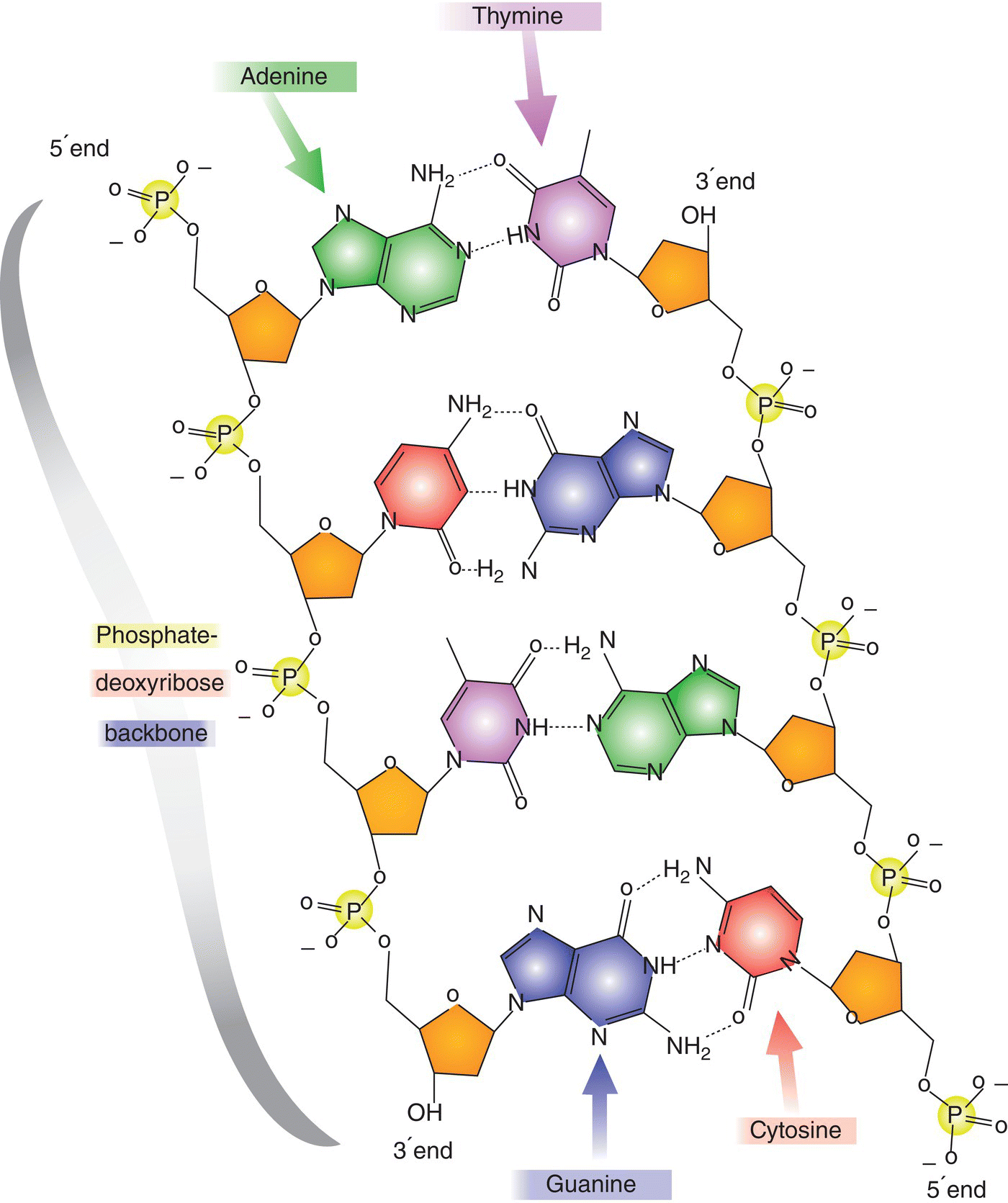
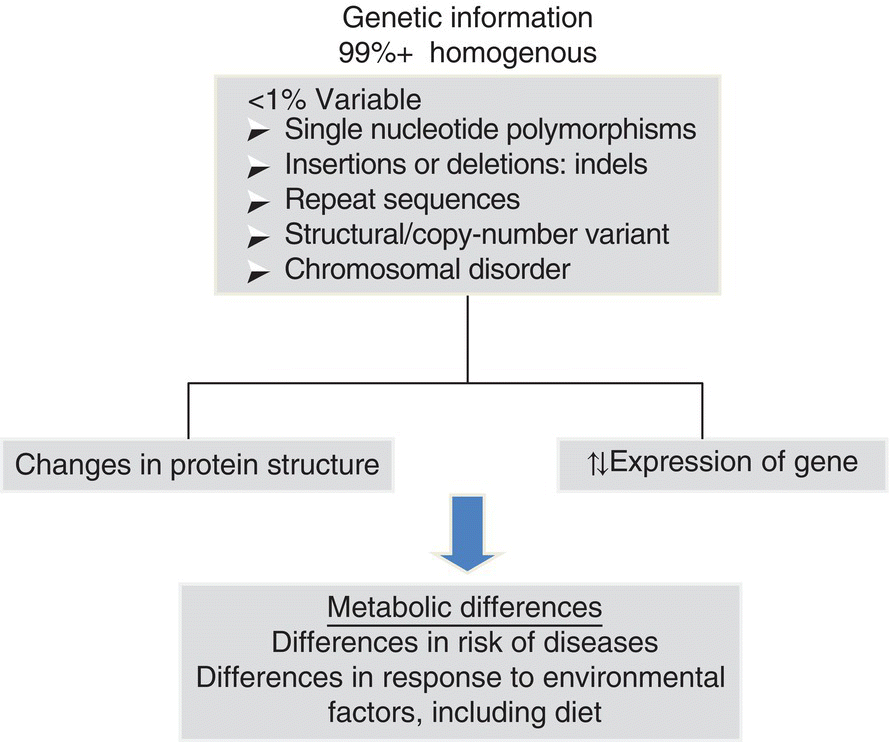
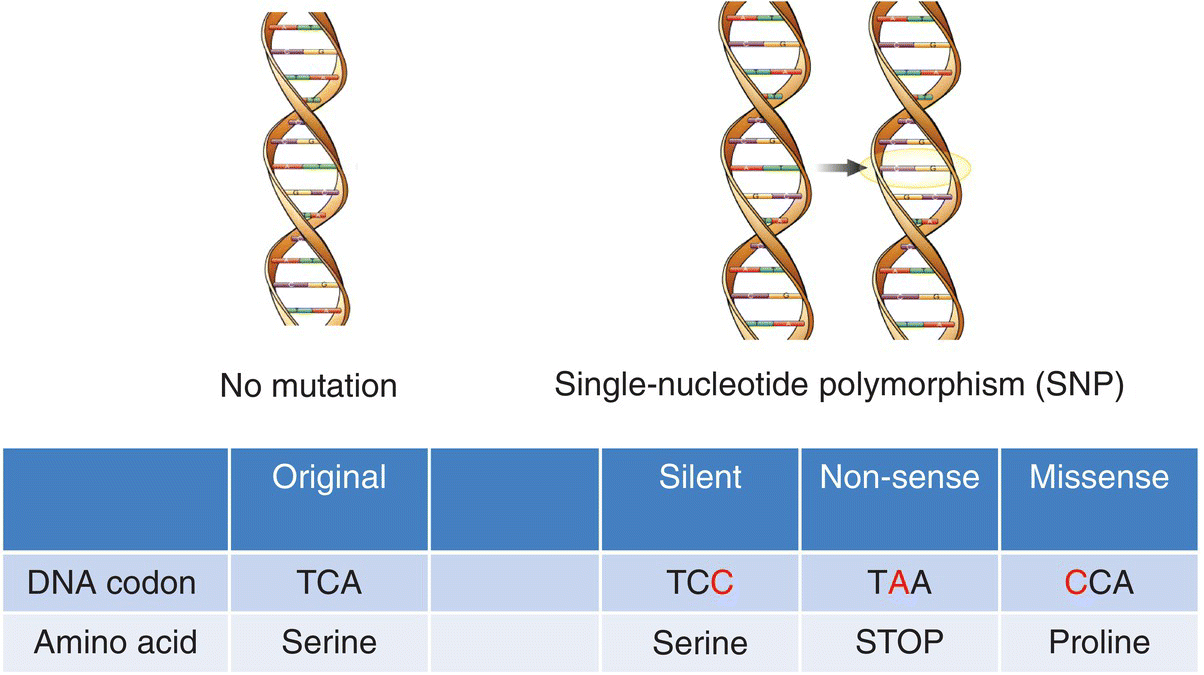
15 Anne Marie Minihane Department of Nutrition, Norwich Medical School, University of East Anglia There is some confusion within the scientific community regarding the terms nutrigenetics and nutrigenomics. Nutrigenetics refers to the impact of genetic variation (genotype) on the response to different diet constituents, whereas nutrigenomics is the effect of dietary components on the expression of genes. Nutrigenomics interactions are the focus of Chapter 13, with the current chapter providing an overview of the various types of genetic variation, scientific approaches and methodologies used to study nutrigenetic interactions, and what is currently known regarding the impact of genetic variability on the risk of disease and response to dietary change. The impact of genotype on the association between diet and disease is a complex one, with genetic variability influencing: Our DNA (deoxyribonucleic acid) contains the information needed to synthesise all the proteins required for human development and function. The first draft of the majority of the sequence of the human genome was published in a Nature article entitled ‘Initial sequencing and analysis of the human genome’ in 2001, with the complete sequence available in 2004. This has subsequently led to the recognition that there is less than 1% genetic variation between individuals. When first published, the availability of the human genome sequence was considered to be a panacea, with genetic information predicted by many commentators to have a major impact on approaches used in the fields of medicine and overall public health. In nutrition it was thought that it would lead to the replacement of generic ‘one size fits all’ dietary guidelines with more efficacious, stratified dietary advice based in part on personal genetic information. However, this has not (as yet) transpired, with only a relatively small fraction of the estimated total genetic contribution to health and response to diet having been identified. This begs the critical question of whether our initial estimates of hereditability (contribution of genotype to phenotype) are inflated, or whether our current methods for the detection of genetic variation or the interpretation of the data have been insensitive or misleading in their approaches. The balance of evidence suggests the latter to be the case, as will be discussed in this chapter. In plants and animals, DNA is organised into 23 pairs of chromosomes within the nucleus of the cell. One member of each pair is inherited from the mother and one from the father. Most DNA molecules are double-stranded helices of two long chains of repeating units called nucleotides. Each nucleotide consists of a sugar and phosphate group (which makes up the backbone of the chain) and a base attached to the sugar (Figure 15.1). The bases are adenine (A), thymine (T), guanine (G) and cytosine (C). Virtually every single cell in the body contains a complete copy of the approximately 3 billion DNA base pairs. A series of three bases encodes for a particular amino acid, with for example ACT, ACC, ACA and ACG coding for threonine, and ATG coding for methionine (Table 15.1). Figure 15.1 Basic nucleotide structure. Table 15.1 DNA codon table. Ala, alanine; Arg, arginine; Asn, Asparagine, Asp, aspartic acid;; Cys, cysteine; Gln, glutamine; Glu, glutamic acid; His, histidine; Ile, isoleucine; Leu, leucine; Lys, lysine; Met, methionine; Phe, phenylalanine; Pro, proline; Ser, serine; Thr, threonine; Try, tryptophan; Tyr, tyrosine; Val, valine DNA was first identified and isolated by Swiss physician and biologist Friedrich Miescher in 1871, with its double-helix structure discovered and published in 1953 by James Watson, Francis Crick and Rosalind Franklin. However, it was not until 2001 when the first output of the Human Genome Project (HGP) was published that information on the sequence of bases that make up DNA was available. The project was officially launched in 1990 through funding from the US National Institutes of Health (NIH) and Department of Energy, and resolved to sequence 95% of the DNA in human cells within 15 years. During the early years of the HGP, the UK’s Wellcome Trust Sanger Institute became a major partner. In 2003, two years ahead of schedule, the HGP had achieved its major targets (http://ghr.nlm.nih.gov/handbook/hgp) as follows: In 2004 the almost complete (99.7%) sequence of human DNA was available. DNA is divided into coding and non-coding regions, with coding regions constituting less than 2% of the total DNA, with the other 98% referred to as junk DNA with unknown biological function. However, unsurprisingly, it is becoming evident through initiatives such as the Encyclopaedia of DNA Elements (ENCODE) Consortium that a large proportion of this non-coding DNA does have essential roles. A mutation is the term used to describe a change in the base sequence and structure of DNA (Figure 15.2). Such mutations can be minor, with a change to only a single base in the DNA sequences referred to as a single-nucleotide polymorphism (SNP), right through to changes in the number of chromosomes. The functional consequence (penetrance) of the genotype varies between no detectable impact on phenotype, through to mutations with complete penetrance where all carriers of the mutation develop the trait or disease. Figure 15.2 Genetic variation and its physiological impact. An SNP occurs every 100–300 bases and SNPs account for over 90% of all genetic variability. When a SNPs occur in the coding region of DNA, they may be silent, non-sense or missense (Figure 15.3). Silent, as the term suggests, has no impact on the amino acid sequence of the protein. A non-sense mutation results in a premature stop codon and a truncated and often non-functional protein. A missense SNP results in an amino acid change in the protein sequence, with the functional consequences dependent on where in the protein the amino acid change has occurred, and also on whether the physiochemical properties of the replacement amino acid are comparable to the original amino acid. Although with a few notable exceptions, such as sickle cell anaemia, SNPs rarely cause the disease, they may increase the risk, as will be discussed later in this chapter. It is well known that SNPs in non-coding regions also have functional consequences, although the metabolic basis for this is less clear. A SNP in the gene promoter region may have an impact on the expression of the gene, whereas those in intergenic regions may affect the translation of the genetic code into a protein, for example by influencing mRNA stability or motility within the cell. Figure 15.3 Categories of single-nucleotide polymorphisms (SNPs). Structural variants, are operationally defined as genomic alterations (deletions, duplications, copy-number variants, insertions, inversions and translocations) that involve segments of DNA that are between 1 Kb and 3 MB, with smaller (<1 Kb) variations that involve DNA insertions or deletions referred to as INDELS. Repeat sequences are a length or nucleotide sequence that is repeated, with the consequences often dependent on the number of repeats. Almost all diseases have some genetic component. A genetic disorder is where the genotype contributes to the aetiology of the disease. For the majority of cases of chronic age-related conditions such as cardiovascular diseases (CVD), Alzheimer’s disease (AD) and osteoporosis, the greater part of the genetic risk is thought to be afforded by multiple low-penetrance SNPs and these disorders are therefore referred to as polygenic. However, a small percentage of the cases of such conditions are caused by high-penetrance single-gene defects (also referred to as Mendalian or monogenic disorders). For example, for AD, mutations in three genes, namely amyloid precursor protein, preselin-1 and preselin-2, have been linked to an early-onset (< 60 years) monogenic form of the disorder, which accounts for less than 5% of all cases. In contrast, the common APOE4 genotype (20–25% of Caucasians) has been genetically linked to the late-onset form of the disease (> 60 years). Individuals with two copies of the APOE4 allele (1–2% of the population) have a 50–90% chance of developing AD by age 85 years, with those with one copy having an approximately 45% chance, compared to a 20% chance in the general population. Furthermore, APOE4 carriers develop AD about a decade earlier than non-APOE4 individuals. However, being an APOE4 carrier is neither necessary nor sufficient to cause the disease. Therefore, this form of AD is considered as polygenic and multifactorial.
Nutrient–Gene Interactions
15.1 Introduction
15.2 DNA and genetic variation
DNA and its discovery timeline
T
C
A
G
T
TTT
Phe
TCT
Ser
TAT
Tyr
TGT
Cys
T
TTC
Phe
TCC
Ser
TAC
Tyr
TGC
Cys
C
TTA
Leu
TCA
Ser
TAA
STOP
TGA
STOP
A
TTG
Leu
TCG
Ser
TAG
STOP
TGG
Trp
G
C
CTT
Leu
CCT
Pro
CAT
His
CGT
Arg
T
CTC
Leu
CCC
Pro
CAC
His
CGC
Arg
C
CTA
Leu
CCA
Pro
CAA
Gln
CGA
Arg
A
CTG
Leu
CCG
Pro
CAG
Gln
CGG
Arg
G
A
ATT
Ile
ACT
Thr
AAT
Asn
AGT
Ser
T
ATC
Ile
ACC
Thr
AAC
Asn
AGC
Ser
C
ATA
Ile
ACA
Thr
AAA
Lys
AGA
Arg
A
ATG
Met
ACG
Thr
AAG
Lys
AGG
Arg
G
G
GTT
Val
GCT
Ala
GAT
Asp
GGT
Gly
T
GTC
Val
GCC
Ala
GAC
Asp
GGC
Gly
C
GTA
Val
GCA
Ala
GAA
Glu
GGA
Gly
A
GTG
Val
GCG
Ala
GAG
Glu
GGG
Gly
G
Genetic variability
Genetics and disease risk
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


