(1)
Institut Bergonié, Bordeaux, France
Abstract
Nuclear receptors constitute a special class of transcription factors that are activated by an extracellular signal. This signal is a hydrophobic substance, such as a steroid hormone, which can cross the plasma membrane and does not require a surface receptor to recognise it and transmit to the nucleus the information it conveys. The intracellular receptor is in charge of this function and of the regulation of target gene expression, after having recognised and bound its ligands. This is the most obvious difference between steroid hormones and polypeptidic hormones, which are recognised by G-protein-coupled receptors (GPCRs) (Chap. 6).
Nuclear receptors are the targets of many drugs aimed at treating metabolic and endocrinal diseases, drugs that can be agonists or antagonists of receptor activity. Two types of frequent cancers are hormone dependent: breast and prostate cancers. The nuclear receptors of oestrogens and androgens are, therefore, of major importance in oncology, because their alterations can participate to oncogenesis and cancer development and because they constitute major therapeutic targets. The hormonal treatments of breast cancer were historically the first ‘targeted therapies’ of cancers, but other nuclear receptors also present a major interest in oncology.
14.1 Structure and Function of Nuclear Receptors
There are about 50 nuclear receptors distributed in three major classes (Table 14.1); one of them (NR3 for nuclear receptor 3) contains especially the steroid hormones receptors: oestrogens (ERα and ERβ, genes ESR1 and ESR2), progesterone (PGR), androgens (AR), glucocorticoids (GR) and mineralocorticoids (MR). Another class (NR1) gathers the receptors of thyroid hormones TRα and TRβ (genes THRA and THRB), vitamin D (VDR), retinoic acid (RARα, RARβ and RARγ; genes RARA, RARB and RARG), oxysterols (LXR, liver X receptor), biliary acids (FXR, farnesoid X receptor), fatty acids and eicosanoids (PPARα, PPARβ and PPARγ, peroxisome proliferator–activated receptor, genes PPARA, PPARD, PPARG) and various xenobiotics (PXR, pregnane X receptor, gene NR1I2; and CAR, constitutive androstane receptor, gene NR1I3). The last class (NR2) only contains the accessory receptors of retinoic acid (RXRα, RXRβ and RXRγ, retinoid X receptors, genes RXRA, RXRB and RXRG). Each class contains in addition some related ‘orphan’ receptors, whose ligand remains unknown, such as ERRα, ERRβ and ERRγ (oestrogen receptor–related receptors). The structure of some ligands of nuclear receptors is presented on Fig. 14.1.
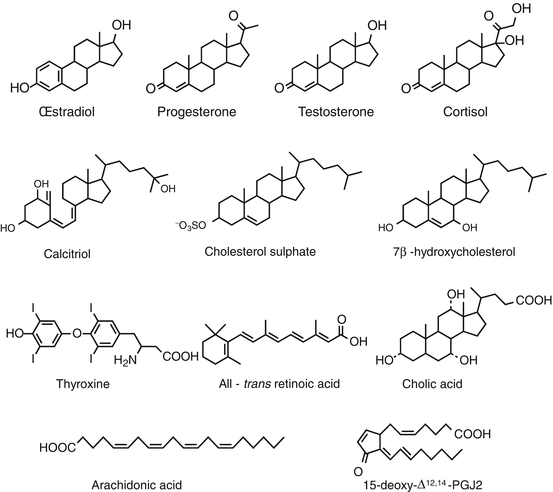
Table 14.1
Main nuclear receptors and their ligands
Usual names | Nomenclature | Main ligands |
|---|---|---|
TRα, TRβ | NR1A1, NR1A2 | Thyroxine, triiodothyronine |
RARα, RARβ, RARγ | NR1B1, NR1B2, NR1B3 | All-trans retinoic acid |
PPARα, PPARβ, PPARγ | NR1C1, NR1C2, NR1C3 | Fatty acids, leukotrienes, prostaglandins |
RORα, RORβ, RORγ | NR1F1, NR1F2, NR1F3 | Cholesterol, cholesterol sulphate |
LXRα, LXRβ | NR1H3, NR1H2 | Oxysterols |
FXRα, FXRβ | NR1H4, NR1H5 | Biliary acids |
VDR | NR1I1 | Vitamin D |
PXR, CAR | NR1I2, NR1I3 | Xenobiotics |
RXRα, RXRβ, RXRγ | NR2B1, NR2B2, NR2B3 | 9-cis-retinoic acid |
ERα and ERβ (ESR1 and ESR2) | NR3A1 et NR3A2 | Oestradiol, tamoxifen |
ERRα, β, γ | NR3B1, NR3B2, NR3B3 | Diethylstilbestrol |
GR | NR3C1 | Cortisol |
MR | NR3C2 | Aldosterone |
PR (PGR) | NR3C3 | Progesterone |
AR | NR3C4 | Testosterone |
AHR | Aromatic hydrocarbons (dioxin) |
Fig. 14.1
Structure of the main ligands of some nuclear receptors. The ligands presented here recognise the following receptors: oestradiol, oestrogen receptor (ER, gene ESR1); progesterone, progesterone receptor (PR, gene PGR); testosterone, androgen receptor (AR); cortisol, glucocorticoid receptor (GR, gene NR3C1); calcitriol, vitamin D receptor (VDR); cholesterol sulphate, RAR-related orphan receptor (RORα, gene RORA); 7β-hydroxycholesterol, liver X-receptor (LXR, gene NR1H2); thyroxine, thyroid hormones receptor (TRα, gene THRA); all-trans retinoic acid (ATRA), retinoic acid receptor (RARα, gene RARA); cholic acid, biliary acid receptor (FXR, gene NR1H4); arachidonic acid and prostaglandin 15-deoxy-Δ12,14-PGJ2, peroxisome proliferator–activated receptor (PPARα, gene PPARA)
Nuclear receptors contain an N-terminal domain of transcription activation in the absence of ligand (AF1, activation function 1), a central domain for DNA binding (DBD, DNA-binding domain), owing to two zinc finger domains, a hinge domain and a C-terminal domain for ligand recognition and binding (LBD, ligand-binding domain), containing the AF2 site for dimerisation and ligand-activated transcription (Fig. 14.2). Depending on the case, nuclear receptors can act as monomers, homodimers or heterodimers and, in this case, always with a RXR receptor. They recognise on DNA a sequence called HRE (hormone–responsive element). This sequence is either repeated in tandem, forming two half-sites separated by a variable number of nucleotides (most frequently 3), or repeated in an inverted, palindromic way, the two half-sites being also separated by a variable number of nucleotides. In both cases, each receptor monomer binds on one of the half-sites.
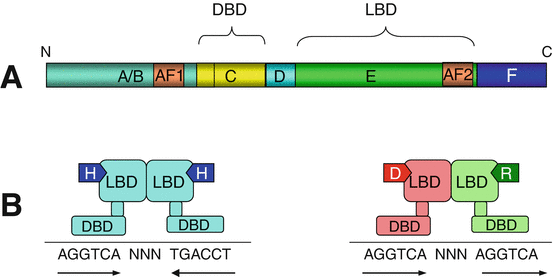
Fig. 14.2
General structure of nuclear receptors. (a) The polypeptidic sequence can be subdivided in A/B, C, D, E and F domains. The A/B domain contains a sequence AF1 (activation function 1) which enables the receptor to exert weak activity in the absence of ligand. C is the DNA-binding domain (DBD) which contains the zinc finger motifs recognising the hormone-responsive elements (HRE) on the target genes. D is a hinge domain allowing the receptor to fold after ligand binding and dimerisation. E is the ligand-binding domain (LBD), which also contains the dimerisation site and the AF2 sequence for ligand-dependent transactivation. (b) The interaction between ligand and receptor leads to the binding of the activated receptor on a DNA sequence. For NR3 receptors (left), there is homodimerisation of the hormone (H)-activated receptor and recognition of a palindromic sequence; for NR1 receptors (right), there is heterodimerisation of the specific receptor (VDR in this example) activated by its ligand (vitamin D) and the common RXR receptor activated by 9-cis-retinoic acid, with recognition of a tandem sequence
Ligand–receptor binding can occur in the cytoplasm; the translocation of the activated receptor to the nucleus is possible thanks to the unmasking of a nuclear localisation sequence (Annex C). In other cases, the receptor is permanently localised in the nucleus; in the absence of ligand, the LBD is bound to a corepressor NCOR (nuclear receptor corepressor); when the LBD has recognised and bound a ligand, the transcription activation domain is activated owing to the intervention of a coactivator NCOA (nuclear receptor coactivator). The function of corepressors and coactivators is to recruit histone-modifying enzymes, which control the degree of methylation and acetylation of histones (Annex B). Histone deacetylation induces a compaction of the chromatin that does not allow target gene transcription, whereas their acetylation facilitates transcription (Fig. 14.3). Nuclear receptors can exert other effects, independently of their transcription function: some of them are able, in the cytoplasm, to activate signalling pathways such as the MAP kinases or the PI3 kinase pathways (Chaps. 2 and 3) or the pathways related to Ca2+ release in the cytosol (Chap. 6).
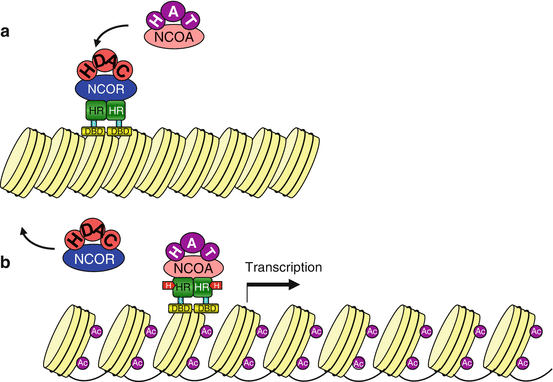
Fig. 14.3
Transcription activation by nuclear receptors. (a) In the absence of ligand, the transcriptional activity of the nuclear receptor is inhibited by a corepressor NCOR, which is associated to histone deacetylase activity HDAC. Chromatin is compact and does not enable transcription. (b) After ligand binding, the receptors can be combined with a coactivator NCOA, which recruits a histone acetyltransferase HAT; chromatin is thus relaxed and the transcription of target genes becomes possible
It is impossible to establish a list, even succinct, of the genes whose transcription is activated by nuclear receptors. Several hundreds of genes harbour, in their promoter sequence, responsive elements specific for one or several activated receptors. The research of target genes can be realised in silico, thanks to algorithms of identification of these promoter sequences over the whole genome, or by analysing the gene expression profiles obtained before and after treatment by a given ligand. The genes transcribed by an activated receptor reflect the characteristics of the cognate hormones, in the fields of cell metabolism, proliferation and differentiation. Oestrogens and androgens, for instance, induce a proliferation of the cells which express oestrogens and androgen receptors, respectively, which can be used by hormone-dependent cancers.
14.2 Steroid Hormones Receptors
Five classes of steroid hormones can activate nuclear receptors: oestrogens, progesterone, androgen, glucocorticoids and mineralocorticoids. In the absence of ligand, these receptors are associated in the cytoplasm to chaperone HSPs (heat shock proteins) which maintain them in inactive state. Ligand binding induces the removal of the HSP, the homodimerisation of the receptor and its translocation into the nucleus. The dimer recognises the target sequence HRE and triggers the transcription of the corresponding genes.
14.2.1 Oestrogens and Progesterone Receptors
The activity of oestrogens on breast cancer growth has been known for a long time, since castration was proposed more than one century ago as breast cancer treatment. Oestrogens favour the proliferation of tissues expressing ERα (NR3A1, gene ESR1), which occurs in 75 % of breast cancers. Oestrogens, used as hormone replacement therapy of menopause, have induced an increase in breast cancer incidence, which is now decreasing, since an international warning on their use has been outspread. The second oestrogen receptor, ERβ (NR3A2, gene ESR2), is a negative regulator of ERα in normal breast tissue, in controlling its transcriptional activity through heterodimerisation, which decreases the transcription rate.
Since the 1970s, a hormonal therapy targeting this relationship between oestrogens and breast cancer growth has been implemented: tamoxifen and its derivatives, under the generic name of SERM (selective oestrogen receptor modulators), display an antagonistic effect to oestradiol, which is in some tissues partially agonistic. After binding to the receptor, they prevent some oestradiol transcriptional effects, but not all. In contrast, SERDs (selective oestrogen receptor downregulators), such as fulvestrant, bind to ERα with the same affinity as oestradiol but without any agonistic activity and inhibit receptor expression at the transcriptional level. In addition to the compounds which bind ERα, another therapeutic approach is available for hormone-dependent breast cancers: the inhibition of aromatase, the enzyme that produces C18 steroids (oestrogens) from C19 steroids (androgens).
Hormone treatments can only be efficient when breast tumour tissue expresses ERα. However, about 30 % of cancers expressing ERα are hormone resistant. For more than 20 years, a biochemical technique evaluating the binding capacity of oestradiol to its receptor in tumour cytosolic extracts was used to restrict tamoxifen prescription to the sole patients whose tumours expressed the receptor. This assay has been replaced by an immunohistochemical technique, more rapid and less expensive, but certainly much less informative. ERβ does not seem to be frequently expressed in breast cancers; anyway, it is not systematically evaluated like ERα. Its expression should be theoretically associated to an absence of hormonal dependency of cancer cells. Progesterone receptor PR (NR3C3, gene PGR) is also routinely evaluated in breast cancers. PR expression is highly correlated to that of ERα, but the two receptors are sometimes dissociated; it appears that the simultaneous presence of both receptors would be required for tamoxifen activity, to which ER+–PR− tumours are less often sensitive than ER+–PR+ tumours.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


