New, next-generation targeted treatment strategies are required to improve outcomes in patients with multiple myeloma (MM). Monoclonal antibodies, cell signaling inhibitors, and selective therapies targeting the bone marrow microenvironment have demonstrated encouraging results with generally manageable toxicity in therapeutic trials of patients with relapsed and refractory disease, each critically informed by preclinical studies. A combination approach of these newer agents with immunomodulators and/or proteasome inhibitors as part of a treatment platform seems to improve the efficacy of anti-MM regimens, even in heavily pretreated patients. Future studies are required to better understand the complex mechanisms of drug resistance in MM.
Key points
- •
New, next-generation targeted treatment strategies are urgently required to improve outcomes in patients with multiple myeloma (MM).
- •
Monoclonal antibodies, cell signaling inhibitors, and selective therapies targeting the bone marrow microenvironment have demonstrated encouraging results with generally manageable toxicity in therapeutic trials of patients with relapsed and refractory disease, each critically informed by preclinical studies.
- •
A combination approach of these newer agents with immunomodulators and/or proteasome inhibitors as part of a treatment platform seems to consistently improve the efficacy of anti-MM regimens, even in heavily pretreated patients.
- •
Future studies continue to be required to better understand the complex mechanisms of drug resistance in MM.
- •
Incorporating molecular correlates to further personalize treatment and to, thus, better integrate these agents into clinical practice is a clear priority.
Introduction
Multiple myeloma (MM) is the second most common hematologic malignancy after non-Hodgkin lymphoma and remains incurable despite major advances in therapy over the last decade, with the use of bortezomib, thalidomide, and lenalidomide as first-generation novel therapies in particular impacting favorably on prognosis. MM remains challenging because of both tumor-specific factors, including adverse mutations that result in both inherent and acquired resistance to therapy, and enhanced tumor survival derived from the surrounding bone marrow microenvironment. Newer targeted treatment strategies are currently under development and show considerable promise in overcoming this resistance; these include second-generation proteasome inhibitors (PIs) (such as carfilzomib), third-generation immunomodulators (specifically pomalidomide), monoclonal antibodies in particular, and other cell signaling inhibitors, as well as specific therapies targeting the bone marrow microenvironment. Some of these agents already have proven highly efficacious in the relapsed and refractory (RR) setting, specifically carfilzomib and pomalidomide; this leads to their recent regulatory approval. This review focuses on novel targeted therapies currently under investigation and in various stages of clinical trials, with approvals pending and/or breakthrough designation assigned to several agents.
Introduction
Multiple myeloma (MM) is the second most common hematologic malignancy after non-Hodgkin lymphoma and remains incurable despite major advances in therapy over the last decade, with the use of bortezomib, thalidomide, and lenalidomide as first-generation novel therapies in particular impacting favorably on prognosis. MM remains challenging because of both tumor-specific factors, including adverse mutations that result in both inherent and acquired resistance to therapy, and enhanced tumor survival derived from the surrounding bone marrow microenvironment. Newer targeted treatment strategies are currently under development and show considerable promise in overcoming this resistance; these include second-generation proteasome inhibitors (PIs) (such as carfilzomib), third-generation immunomodulators (specifically pomalidomide), monoclonal antibodies in particular, and other cell signaling inhibitors, as well as specific therapies targeting the bone marrow microenvironment. Some of these agents already have proven highly efficacious in the relapsed and refractory (RR) setting, specifically carfilzomib and pomalidomide; this leads to their recent regulatory approval. This review focuses on novel targeted therapies currently under investigation and in various stages of clinical trials, with approvals pending and/or breakthrough designation assigned to several agents.
Second-generation proteosome inhibitors
Ixazomib (MLN9708)
MLN9708 is a dipeptidilic boronic acid that immediately hydrolyzes to MLN2238, an active form, on exposure to aqueous solutions. MLN2238 reversibly and selectively inhibits the 20S proteasome. It has a 6-fold faster dissociation half-life and greater tissue penetration as compared with bortezomib ( Fig. 1 ). In a xenograft model, MLN2238 showed significantly longer survival in tumor-bearing mice treated with MLN2238 than mice receiving bortezomib. MLN2238 was found to be active even in bortezomib-resistant cells. MLN2238 can synergize with lenalidomide, vorinostat, and/or dexamethasone in combination. Importantly, MLN9708 did not demonstrate significant inhibition of neuronal cell survival, which may explain the lack of major peripheral neuropathy (PN) seen so far with this PI.
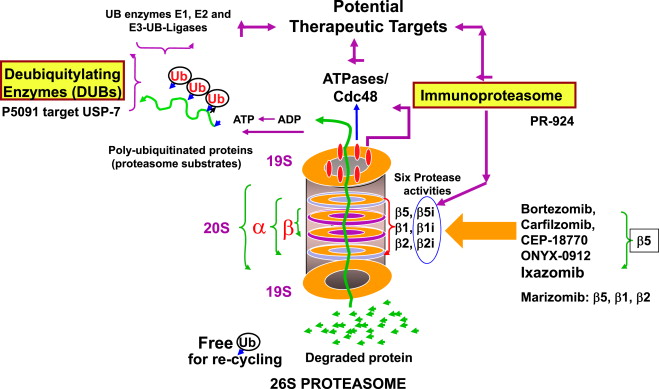
MLN9708 was the first oral PI to be incorporated into clinical trials. Several phase I studies have evaluated the safety of MLN9708 using both oral and intravenous (IV) routes of administration. Data from these studies have demonstrated linear pharmacokinetics, regardless of administration route.
Two phase I studies, one using weekly dosing and the other using biweekly dosing, have evaluated the oral administration of MLN9708 as a single agent in heavily pretreated patients with RR MM previously exposed to PIs. In the 41 evaluable patients receiving weekly dosing, responses included 1 very good partial response (VGPR), 5 partial responses (PR), 1 minimal response (MR), and 15 with stable disease (SD). In the 46 evaluable patients receiving biweekly dosing, 6 patients achieved PR or more, including 1 stringent complete remission (sCR) and 5 PR.
The most common all-grades adverse events (AEs) included fatigue (30%–40%), thrombocytopenia (30%–40%), nausea (30%), diarrhea (25%), vomiting (20%), as well as rash and neutropenia. Drug-related PN was minimal at 4% to 8%, and none were grade 3 or more. Toxicities proved generally manageable with dose reduction and supportive care, and tolerability overall was considered favorable.
Preliminary data from phase I/II studies of once-weekly and biweekly oral MLN9708 in combination with lenalidomide and dexamethasone in patients with newly diagnosed myeloma were recently reported. Drug-related AEs were similar to the phase I studies, although more frequent with twice-weekly administration. Among evaluable patients who received biweekly dosing, 95% achieved PR or better, with a 27% CR/sCR rate. The depth of response increased over the course of treatment. The median time to best response was 1.96 months. MLN9708 in combination with lenalidomide and dexamethasone is currently being investigated in a phase III trial in newly diagnosed patients as well as having been recently completed in patients with relapsed disease, with results expected next year. The benefit of maintenance therapy with this agent is also being evaluated both as a single agent and in combination with lenalidomide.
Oprozomib (ONX 0912)
Oprozomib (ONX 0192) is an epoxyketone PI and is an orally available analogue of carfilzomib. Both carfilzomib and ONX 0912 are irreversible PIs that selectively inhibit the chymotrypsin-like (CT-L) subunit (β5 and β5i) and, thus, have minimal off-target effects (see Fig. 1 ). This PI has shown a significant anti-MM response in vitro and in vivo studies. ONX 0912 is currently being evaluated in phase I and II dose-escalating studies. Reported adverse effects are mainly gastrointestinal and similar to observations in preclinical animal models. Although promising activity has been seen, serious gastrointestinal toxicity has been reported; concerted efforts to improve tolerability are underway.
NPI-0052 (Marizomib)
NPI-0052 (Marizomib) is part of a unique class of PIs, in that it is a natural β-lactone compound that can irreversibly inhibit CT-L, trypsin-like (T-L), and caspase-like (C-L) proteasomal activities in vitro and in vivo (see Fig. 1 ). Through its covalent binding of all 3β subunits, NPI-0052 demonstrates comparable or even greater proteasome inhibition compared with bortezomib and carfilzomib in preclinical models. NPI-0052 is also highly synergistic with either lenalidomide or pomalidomide in vitro and overcomes bortezomib resistance preclinically.
Subsequent results from a dose-escalating study in patients with RR MM with twice-weekly IV NPI-0052 has been reported, with 20% (3 of 15) of bortezomib-resistant patients achieving PR, and 73% of all evaluable patients (n = 22) achieving at least SD. Reported dose-limiting toxicities (DLTs) included predominantly mild to moderate hallucinations, cognitive changes, and loss of balance, which were transient and reversible with dose reduction. Importantly, there was no evidence of treatment-emergent PN or significant myelosuppression. A twice-weekly regimen is currently being investigated, and combination studies with pomalidomide and dexamethasone are underway.
Cell signaling agents
Histone Deacetylases Inhibitors
Epigenetic modification plays a diverse role in both physiologic and pathologic cellular processes. Acetylation, one of the most frequent alterations in epigenetics, serves as a key player in the regulation of gene expression by altering chromatin structure without modifying the underlying DNA. Acetylation is tightly regulated by 2 opposing enzymes: histone deacetylases (HDACs) and histone acetyltransferases (HATs). Although hyperacetylation of the histone NH2 tail by HATs results in open chromatin and gene expression, HDACs have shown to have a repressive effect on transcription by mediating a closed chromatin conformation. Transcriptional repression by HDACs is implicated in carcinogenesis, making them a promising therapeutic target. HDACs also act on many other nonhistone substrate proteins involved in the modulation of transcription.
HDAC inhibitors bind to the catalytic sites of HDACs, preventing accessibility of transcription factors to promoter regions and also upregulate negative cell cycle modulators of the G1 phase, such as p21 WAF1 and p27 Kip1 . HDAC inhibitors can be categorized as class–specific inhibitors or as pan-deacetylase (pan-DAC) inhibitors, the latter denoting activity against both class I and II recombinant HDACs.
Panobinostat
Panobinostat is a potent pan-DAC inhibitor, which has potent inhibitory activity at low nanomolar concentrations against all class I, II, and IV HDACs ( Fig. 2 ). Panobinostat leads to the acetylation of lysine residues across intracellular targets. In preclinical studies, panobinostat had an antimyeloma effect using in vitro and in vivo myeloma models. Based on this data, a phase II study of oral panobinostat as monotherapy in heavily pretreated patients with RR MM was investigated. Patients had at least 2 prior treatments, including bortezomib, thalidomide, and lenalidomide. Panobinostat was administered at a dosage of 20 mg 3 times weekly out of a 21-day treatment cycle. Overall, one PR and one MR were observed out of 38 evaluable patients; however, these responses were maintained for 19 and 28 months, respectively. All grade AEs reported included gastrointestinal toxicity (80.0%) and hematologic effects, which included grade 3/4 neutropenia (31.6%), thrombocytopenia (26.3%), and anemia (18.4%).
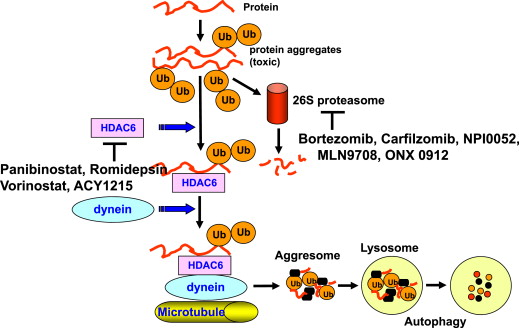
Panobinostat was next evaluated in combination with bortezomib in patients with RR MM, based on preclinical studies, which demonstrated synergy. Specifically, in a phase Ib study of panobinostat and bortezomib, clinical responses were observed (including complete responses) in patients with bortezomib-refractory MM; toxicity proved manageable.
PANORAMA 2 was a multicenter phase II study examining the combination of oral panobinostat with bortezomib and dexamethasone in 55 heavily pretreated patients with relapsed and bortezomib-refractory MM. The overall response rate (ORR) was 34.5% (including 1 near PR and 18 PRs), with 10 patients achieving an MR, resulting in a clinical benefit rate (CBR) of 52.7%. Among patients with adverse cytogenetics (n = 14), the ORR was 43% and the CBR was 71%. The median progression-free survival (PFS) was 5.4 months. Common grade 3/4 AEs included thrombocytopenia (63.6%), fatigue (20.0%), and diarrhea (20.0%).
Based on these encouraging data, a phase III clinical trial was performed. PANORAMA 1 was an international, randomized, double-blinded study of panobinostat (vs placebo) in combination with bortezomib and dexamethasone in patients with RR MM. A total of 768 patients were randomized. Preliminary results demonstrated an ORR of 61% in the panobinostat arm versus 55% in the placebo arm, with an near CR (nCR)/CR rate of 28% versus 16% ( P = .00006), respectively. The median PFS was 12.0 months versus 8.1 months ( P <.0001; hazard ratio 0.63, 95% confidence interval [0.52, 0.76]) in favor of the panobinostat arm. Common grade 3/4 AEs in the panobinostat versus placebo arms included thrombocytopenia (67% vs 31%), neutropenia (35% vs 11%), and diarrhea (26% vs 8%), which were generally manageable with dose reduction and/or supportive care. Given the PFS benefit of 4 months in favor of panobinostat and its activity in high-risk groups in particular, as well as the encouraging quality of response differences seen, regulatory approval is anticipated this year.
Vorinostat
Similar to panobinostat, vorinostat is also a pan-DAC inhibitor in the hydroxamic class (see Fig. 2 ). Mitsiades and colleagues demonstrated in vitro antimyeloma activity when MM cells were irreversibly committed to cell death after hours of incubation with vorinostat. Using microarray analyses, vorinostat-induced apoptosis was associated with suppression of genes mediating cytokine-driven proliferation and survival, drug-resistance, DNA synthesis/repair, and proteasome function. In MM cell lines, vorinostat successfully induced apoptosis in all tumor cells with increased levels of proapoptotic protein levels of p21 and p53. Vorinostat also inhibited the secretion of interleukin 6 (IL-6) produced by bone marrow stromal cells (BMSCs), suggesting that HDAC inhibitors can overcome cell adhesion–mediated drug resistance (CAM-DR). An ongoing phase I trial examined the combination of vorinostat with lenalidomide, bortezomib, and dexamethasone in patients with newly diagnosed MM. Vorinostat was administered orally at 100, 200, 300, or 400 mg daily on days 1 to 14 of each cycle. Thirty patients were enrolled with an ORR (PR or better) of 100%, with a VGPR or better of 52%, and a CR rate of 28%. At a median follow-up of 11.5 months (range 1–31 months), there has been only one patient with progressive disease. Similarly encouraging results with vorinostat in combination with lenalidomide in RR MM were seen in a phase I combination study with an ORR of 47% and favorable tolerability. In contrast, efforts with vorinostat combined with bortezomib proved challenging; ultimately, the prospective international randomized phase III trial of vorinostat combined with bortezomib versus bortezomib alone failed to demonstrate a meaningful clinical benefit. This finding was despite significantly higher rates of response in favor of the combination and largely because of the excessive toxicity seen with the particular dose and schedule of vorinostat used, as well as the absence of dexamethasone use, resulting in only a minimal PFS improvement of less than a month seen between the doublet and the monotherapy control.
ACY-1215 (Rocilinostat)
HDAC6 plays an important role in the breakdown of ubiquitinated proteins and in the formation of perinuclear aggresomes. Blocking HDAC6 activity results in the accumulation of polyubiquitinated proteins, which, in turn, induces cell stress followed by apoptosis. HDAC6 inhibition markedly enhances the action of PIs, making HDAC6 a promising novel target for this approach as well as with other combinations. Furthermore, the more selective inhibition of HDAC6 may reduce the off-target toxicity previously seen with pan-HDAC inhibitors.
ACY-1215 is a novel, selective HDAC6 inhibitor that is orally available (see Fig. 2 ). Santo and colleagues evaluated the action of ACY-1215 alone and in combination with bortezomib in the preclinical setting. In this study, the combination of both proteasome and HDAC6 inhibition lead to synergistic cytotoxicity, resulting in apoptosis of MM cells by activation of the caspase pathway. In vivo experiments using 2 xenograft severe combined immunodeficiency (SCID) mouse models, confirmed the anti-MM effects of ACY-1215 when combined with bortezomib. The mouse treated with both agents experienced a significantly prolonged overall survival and delayed tumor growth. This study prompted the rationale to use ACY-1215 in clinical trials.
ACY-100 is a single arm, open-label, dose-escalation trial using ACY-1215 in patients with RR MM as monotherapy (phase Ia) and in combination with bortezomib (phase Ib) followed by a phase II extension. ACY-1215 was given orally on days 1 to 5 and 8 to 12 of a 21-day cycle. Most AEs were grade 1 to 2, whereas 2 patients had grade 3 AEs (anemia and neutropenia). No DLTs were observed. Six patients had SD as their best response.
In the combination cohort, treatment-related AEs were mainly low grade, with the majority not considered related to ACY-1215. The first cohort was expanded because of a DLT of asymptomatic increase in amylase, but no other DLTs have been observed. Grade 3 to 4 AEs included asymptomatic elevated amylase, thrombocytopenia, anemia, stomach cramps, and an increase in creatinine. Of the 16 evaluable patients, 1 VGPR, 2 PR, 1 MR, and 5 SD were reported. Of the patients who were previously refractory to bortezomib, the best outcome at the time of presentation was 1 VGPR, 1 MR, and 4 SD, with recent updates suggesting greater ORR with more time on therapy.
Based on synergy observed between ACY-1215 and lenalidomide in preclinical studies, a phase I trial investigating this combination treatment regimen in patients with RR MM is being carried out. In part A, patients were treated with escalating doses of oral ACY-1215 on days 1 to 5 and 8 to 12 of a 28-day cycle, with lenalidomide 25 mg on day 1 to 21 and dexamethasone 40 mg weekly. Most treatment-related events were low grade and included fatigue (43%), upper respiratory infection (36%), anemia and peripheral edema (21% each), neutropenia (29%) and muscle spasms (21%). There were 9 grade 3 to 4 events in 6 patients, which were primarily hematologic. Nine patients (69%) achieved responses of PR or greater, including 1 CR, 4 VGPR, and 3 PR. Of the 6 patients who were previously refractory to lenalidomide, the best responses included 1 PR, 1 VGPR, 2 MR, and 2 SD. Future studies are now evaluating the combination of ACY-1215 with pomalidomide and with whom even greater preclinical activity is seen; the all-oral 3-drug platform of AC1215, pomalidomide, and dexamethasone is, therefore, hoped to be a particularly important triplet going forward.
Heat-Shock Protein 90 Inhibitors
Heat-shock protein 90 (Hsp90) regulates cellular trafficking by facilitating the 3-dimensional folding of intracellular proteins implicated in cell proliferation and drug resistance. In tumors, Hsp90s are an important target, as they act as a chaperone to mutated or overexpressed proteins that promote cell survival. In a phase I/II clinical trial, patients with RR MM were administered the hsp90 inhibitor tanespimycin (100–340 mg/m 2 ) and bortezomib (0.7–1.3 mg/m 2 IV) on days 1, 4, 8, and 11 in each 21-day cycle. Among the 67 evaluable patients, there were 2 (3%) complete responses and 8 (12%) PRs, for an ORR of 27%, including 8 (12%) MRs. The most common AEs were diarrhea (60%), nausea (49%), fatigue (49%), thrombocytopenia (40%), transient elevations in AST (28%) and dizziness (28%). Most toxicities were grade 1 or grade 2. There was no reported grade 3/4 peripheral neuropathy. Unfortunately, the tanespimycin program had to be closed prematurely because of insurmountable difficulties in producing adequate and high-quality amounts of the drug substance. Other studies of Hsp90 inhibition are ongoing, and early results show some promise (eg, AUY922).
Phosphoinosiide 3-kinase/Akt/Mammalian Target of Rapamycin Pathway Inhibitors
Akt modulates the phosphorylation of several downstream substrates involved in cellular growth and survival. One of the most studied downstream protein kinases is the mammalian target of rapamycin, which has been implicated in the pathogenesis of several different cancers. In MM, the phosphoinosiide 3-kinase (PI3K)/Akt pathway is overactive, thus inhibiting apoptosis and allowing for clonal cell expansion. Hsu and colleagues demonstrated, using immunohistochemistry, that Akt is frequently activated in MM cells and the frequency is directly proportional to the disease stage. Interruption of the Akt pathway resulted in inhibition of MM cell growth in vitro.
Perifosine is a biologically available alkylphospholipid that inhibits the Akt pathway and, thus, promotes apoptosis in MM cells. In preclinical studies, baseline phosphorylation of Akt in MM cells was completely inhibited by perifosine in a time- and dose-dependent manner. Perifosine was also successful in inducing apoptosis even in MM cells adherent to BMSCs. Perifosine was found to enhance the cytotoxic effects of novel agents, such as bortezomib. Taken together, these data provided the rationale for clinical trials using Akt pathway inhibitors in the setting of patients with RR MM.
A phase I multicenter single-arm study was carried out looking at escalating doses of perifosine 50 to 100 mg in combination with lenalidomide plus dexamethasone 20 to 40 mg weekly. The most common AEs were grade 1 to 2 fatigue (48%) and diarrhea (45%), and grade 3 to 4 neutropenia (26%), hypophosphatemia (23%), thrombocytopenia (16%), and leucopenia (13%). MR or better was attained in 73% of evaluable patients, including 50% with a PR or better.
In a multicenter phase I/II study, perifosine was combined with bortezomib with or without dexamethasone in 84 patients with RR MM. All patients were heavily pretreated, and many were resistant to bortezomib. The ORR (MR or better) was 41%, including an ORR of 32% in bortezomib-refractory patients. The median PFS was 6.4 months, with a median overall survival of 25 months. Treatment was well tolerated. Common treatment-related grade 1 and 2 AEs included nausea (63%), diarrhea (57%), fatigue (43%), musculoskeletal pain (42%), anorexia, and upper respiratory tract infections (33% each). All AEs were manageable with supportive care and dose reductions. Grade 3 or more toxicities included thrombocytopenia (23%), neutropenia (15%), anemia (14%), and pneumonia (12%). Unfortunately, the pivotal prospective phase III study of perifosine, bortezomib, and dexamethasone versus placebo, bortezomib, and dexamethasone was closed prematurely as a result of resource constraints, slow accrual, and equivocal findings at interim analysis. Other studies with more potent Akt inhibitors are showing considerable promise (eg, GSK2110183). Most recently, Mimura and colleagues also demonstrated anti-MM activities of a novel allosteric inhibitor TAS-117 alone and in combination with bortezomib.
BET Bromodomain Inhibitors
Myc plays a key role in the pathogenesis of many human cancers, including MM. We have yet to discover therapeutic approaches to modulate the function of the c-Myc oncoprotein. Bromodomains are important recognition domains of coactivator proteins implicated in the initiation of transcription. Disruption of the bromodomains will interfere with signal transduction and, ultimately, will inhibit the transcription of the Myc oncoprotein. JQ1 is a selective small-molecule BET bromodomain inhibitor that downregulates Myc transcription and the expression of other Myc-dependent target genes.
Antiproliferative activity of JQ1 was assessed using in vitro and xenograft models. MM cell proliferation was uniformly inhibited by JQ1. These samples included several cell lines resistant to Food and Drug Administration–approved agents. In primary cells isolated from a patient with RR MM, JQ1 treatment resulted in a time-dependent suppression of c-Myc expression. It is hoped that clinical efficacy of the JQ1 inhibitor will be confirmed in human studies; clinical trials are now underway, with combination studies incorporating lenalidomide due to commence this year.
Deubiquitinating Enzyme Inhibitors
Ubiquitin regulates the degradation of proteins via proteasomes and lysosomes and modulates protein-protein interactions. Deubiquitinating enzymes (DUBs) are a group of proteases that cleave the bond between ubiquitin and its substrate protein (see Fig. 1 ). Inhibition of DUBs or proteasome results in the accumulation of ubiquitinated proteins. The novel regulatory particle b-AP15 selectively blocks deubiquitinating activity without inhibiting proteasome activity. In preclinical studies, b-AP15 was shown to decrease viability in bortezomib-resistant MM cell lines and patient MM cells, even in the presence of BMSCs ; b-AP15 demonstrated good tolerability in human MM xenograft models. Combining b-AP15 with lenalidomide or dexamethasone induced synergistic anti-MM activity. DUB inhibitors will need to be further investigated as potential therapeutic agents to improve clinical outcomes in MM. To this end, clinical studies are planned and are expected to begin shortly.
Wnt, Hedgehog, Notch Inhibitors
Wnt
Wnt proteins are glycoproteins that serve as ligands to transmembrane receptors. Abnormal Wnt signaling has been described in MM. Dickkopf 1 (DKK1) is a soluble antagonist of the Wnt pathway that is overexpressed by plasma cells in patients with osteolytic lesions. The overexpression of DKK1 blocks the differentiation of osteoblasts and, thus, inhibits the formation of bone. BHQ880 is a fully human neutralizing antibody targeting DKK1. Preclinical studies have demonstrated that BHQ880 reduces IL-6 secretion and promotes osteoblastogenesis in vitro and in mouse models. Preliminary data from a phase I study in patients with RR MM receiving both zoledronic acid and BHQ880 demonstrated an increase in bone density in some patients. An open-label, multicenter, single-arm, phase II study designed to evaluate the safety and antimyeloma activity of BHQ880 in patients with high-risk smoldering MM (SMM) has recently been completed with excellent tolerability and some suggestion of activity seen.
Notch
The Notch pathway regulates cell proliferation, cellular differentiation, and programmed cell death. It is implicated in the pathophysiology of multiple hematologic malignancies, including MM. MRK003 is a γ -secretase inhibitor that has demonstrated in vitro anti-MM activity by blocking the Notch pathway. There may be preclinical evidence that it also increases sensitivity to bortezomib, and clinical evaluation is under consideration.
Hedgehog
The Hedgehog (Hh) pathway is necessary for cell growth and differentiation; its deregulation has been associated with several cancers, including MM. NVP-LDE225 is a novel antagonist currently in development and has demonstrated an anti-MM activity in vitro by the downregulation of the Hh pathway. Clinical studies in MM are also being considered and should soon be underway.
Kinesin Spindle Protein Inhibitors
Kinesin spindle protein (Ksp) inhibitor (ARRY-520) is a synthetic antimitotic agent that induces the death of actively dividing cells by targeting the Ksp, an essential component of mitosis. Ksp is a microtubule protein that is necessary in the formation of spindles. ARRY-520 has shown promising activity not only as a single agent, but also in combination with bortezomib or lenalidomide in preclinical studies using MM cell lines and xenograft models. These data also demonstrated the ability of ARRY-520 to downregulate mcl-1, a known driver in the development of dexamethasone resistance. Based on phase I clinical studies in the RR setting, a phase II study with ARRY-520 as a single agent and in combination with dexamethasone was carried out. All patients had previously received an immunomodulator; 90% had received prior bortezomib, and 78% had prior autologous stem cell transplant. The most commonly reported treatment-emergent AEs were thrombocytopenia, anemia, neutropenia, and fatigue. The most frequent grade 3 or 4 AEs included neutropenia (62%) and thrombocytopenia (57%). Of the 32 patients in the single-agent arm, an MR or greater was observed in 6 patients (19%), 5 of which were PRs. Among patients who were bortezomib and lenalidomide refractory, a 15% ORR (≥MR) was observed. Patients who received combination ARRY-520 and dexamethasone, the ORR was 28% (5 of 18), with 4 patients achieving a PR or greater. There was no association between ARRY-520 and the development of peripheral neuropathy. Further evaluation of ARRY-520 in combination with other novel agents, such as bortezomib and carfilzomib, are underway.
Chromosome Region Maintenance 1
Chromosome region maintenance 1 (CMR1) is a nuclear export protein used to transfer proteins with leucine-rich nuclear sequences from the nucleus to the cytoplasm. This shuttling system is very tightly regulated, given that its cargo includes tumor suppressor proteins, such as p53.
The overexpression of CRM1 is responsible for the abnormal cellular localization of tumor suppressive proteins and has been implicated in the development of certain cancers. Tai and colleagues demonstrated that CRM1 is highly expressed in patients with MM, including those who are refractory to bortezomib. The overexpression of CRM1 was also correlated with lytic bone disease and a shorter survival. Irreversible selective inhibitors of nuclear export (SINE) targeting CRM1 (KPT-185, KPT-330) induced cell death in MM cells by the accumulation of CRM1 cargo tumor suppressor proteins, even in the presence of BMSCs or osteoclasts. In mice models with MM bone lesions, SINEs successfully inhibited bone lysis by impairing osteoclastogenesis and bone resorption by blocking the nuclear factor–κB pathway. These results are convincing that CRM1 is an important therapeutic target and requires further investigation in human studies.
Related posts:
 Treatment of Transplant-Eligible Patients with Multiple Myeloma in 2014
Maintenance Therapy for Multiple Myeloma
Treatment of Transplant-Eligible Patients with Multiple Myeloma in 2014
Maintenance Therapy for Multiple Myeloma
 Monoclonal Gammopathy of Undetermined Significance and Smoldering Multiple Myeloma
Relapsed and Refractory Multiple Myeloma
Novel Targeted Agents in the Treatment of Multiple Myeloma
Immunotherapy Strategies in Multiple Myeloma
Monoclonal Gammopathy of Undetermined Significance and Smoldering Multiple Myeloma
Relapsed and Refractory Multiple Myeloma
Novel Targeted Agents in the Treatment of Multiple Myeloma
Immunotherapy Strategies in Multiple Myeloma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


