Fig. 2.1
Short-term protection observed at 4 weeks against loss of cancellous bone mass in rats achieving reduced energy availability with exercise and dietary energy restriction (ER) disappears after 12 weeks. Decline in volumetric bone mineral density (vBMD) at 12 weeks in ER-exercised animals is still half that observed in ER-sedentary (SED) rats. Delta values with the same letter superscript are not significantly different; *p < 0.05 vs. baseline value on day 0. Data from Swift et al. [21]
Estrogen Receptor-Alpha and Mechanotransduction in Bone
Recently, a fascinating new line of research has examined the effect of reduced circulating estrogens may have on bone’s ability to adapt to mechanical strains and exercise. It has long been known that bone structure adapts to the typical loading patterns to which it is subjected. Current recommendations focus on weight-bearing exercise and resistance training as effective means of increasing bone mass across most of the life-span. However, bone is less responsive to even optimal exercise modes with increasing age [38]. Interestingly, the sensitivity of bone’s response to loading is also reduced in individuals with estrogen deficiency, including postmenopausal women, amenorrheic athletes, and men with progressive estrogen deficiency [39]. This observation led to the hypothesis that low serum estrogen or a closely related factor is responsible for this diminished sensitivity of bone to mechanical loading; key candidates were the intracellular estrogen receptors, of which there are two: ERα or estrogen receptor-β (ERβ). ERα became a likely candidate for this role as it is expressed in osteoblasts, osteoclasts, and osteocytes [39]. Also, it has been previously shown in humans as well as animals that the estrogen receptor expression varies with levels of circulating estrogen [40, 41]. Therefore, a decline in circulating estrogen would lead to fewer estrogen receptors expressed in bone cells. If these receptors have any influence on key signaling pathways important to the regulation of bone formation activity in osteoblasts, an individual with low estrogen status would be predicted to have an attenuated bone response to exercise.
The first clue regarding ERα’s response in mechanical loading of bone was provided by in vitro experiments in which osteoblast-like cells were exposed to mechanical strain and treated with the estrogen receptor modulators ICI-182,780 and tamoxifen [42]. These two compounds, both estrogen receptor antagonists, eliminated or reduced osteoblast proliferation to the mechanical strain, suggesting that bone’s adaptive response to mechanical strain is mediated through a mechanism which involves ERα. To confirm these findings in vivo, Lee et al. [43] studied the response to mechanical loading of bone in ERα-null mice. This commonly used model provides axial loading of the ulna using a servomotor device to cyclically deform the bone in an anesthetized animal; the key advantage of this model is to provide very precise control over the loading signal delivered (strain magnitude and strain distribution), but it does not involve muscle contraction or the integrated physiological response to voluntary exercise. While wild-type mice exhibited an 8 % increase in cortical area at the midshaft ulna, the response in ERα-null mice was nearly fourfold lower (2.4 % gain in bone area) (see Fig. 2.2). Primary osteoblast cultures from the ERα knockout mice exhibited no proliferative response when exposed to strain in vitro, while cells derived from wild-type littermates increased in number by 58 %. These data demonstrate some critical role for ERα activity in the adaptive response of bone to strain-related signals. The loss of bone and reduction in ability to respond to exercise in estrogen deficiency could be explained through the estrogen receptor’s role in adaptation to loading.
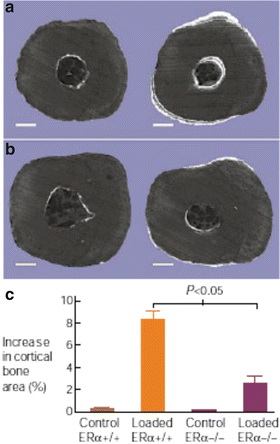
Fig. 2.2
Mechanotransduction is impaired in the absence of estrogen receptor-alpha (ERα). Transverse sections of unloaded control (left) and loaded (right) ulnae from (a) ERα wild-type and (b) ERα knockout mice. White fluorochrome label on bone surfaces represents newly formed bone. (c) Quantification of increased cortical bone area in the loaded versus control wild-type and knockout mice. From Lee et al. [43]
The role of ERβ is not as well established as that of ERα, but it, too, might have a role in the loading response of bone. The fact that there are two estrogen receptors present in bone has made determining the role of each individual receptor a greater challenge. ERβ is also present in osteoblasts, but early research was inconclusive as to its impact on the loading response. With in vivo loading of ERα knockout mice and ERβ knockout mice, similar changes in bone are seen when the estrogen receptors are individually silenced. ERβ knockout mice exhibit only half the increase in cortical area due to periosteal expansion as do ERβ-intact (wild-type) mice [44]. This impact of the missing ERβ on the cortical bone gain is about half of that observed in ERα-null mice (nearly a fourfold decrease) [43], but it appears that the absence of either ERα or ERβ results in attenuation of the osteogenic response to loading.
To complicate this story, more recent research demonstrates that there may be differential roles for ERα and ERβ in males and females. With ERα or ERβ deleted, the different effects of either reduced loading on tibial bone (via sciatic neurectomy) or increased loading were investigated in male and female mice [45]. In both genders ERα-null, but not ERβ-null, mice exhibited an attenuated loss of cancellous bone after prolonged disuse, as compared to their wild-type counterparts. Those female mice lacking ERα had a diminished osteogenic response to loading on cortical bone but, interestingly, no effect was observed for cancellous bone surfaces. By contrast, ERα knockout males had a greater osteogenic response (vs. wild-type littermates) to increased loading on both cortical and cancellous bone surfaces. Deletion of ERβ, on the other hand, resulted in an increased osteogenic response on cortical bone surfaces in both male and female mice, suggesting a tonic inhibition of periosteal osteoblast response to loading in the intact animal [45]. The precise role of these two estrogen receptors in mechanotransduction may be more complex than initially thought, given these gender-specific responses.
Another intriguing question is if mechanical loading or exercise is capable of independently affecting estrogen receptor expression in bone cells. Osteoblast and osteocyte expression levels of ER-α protein are 6- and 26-fold greater, respectively, in female rats after 1 week (3 bouts) of loading achieved by active muscle contraction (see Fig. 2.3) [46]. Interestingly, 12 weeks of energy restriction preceding the week of imposed loading, which usually diminishes circulating estrogen, does not diminish these increases in ER-α expression, but does blunt the bone formation response to mechanical loading. Hence, in this case, the anabolic response of osteoblasts to exercise does not appear to be caused by a down-regulation in ER-α protein in osteoblasts or osteocytes.
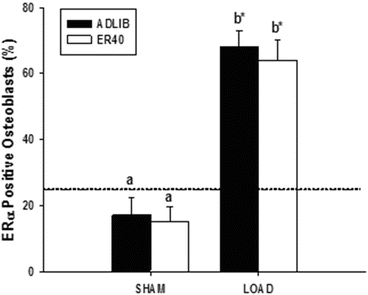
Fig. 2.3
Short-term mechanical loading (LOAD) upregulates ER-α expression in (a) osteoblasts in the distal femur metaphysis, even in animals subjected to 12 weeks of 40 % reduction in energy intake (ER-40). Dashed line denotes baseline control (BC) animals’ values. Those groups not sharing the same letter are significantly different from each other (p < 0.05). *p < 0.05 versus baseline control mean value. Data from Swift et al. [46]
While more research needs to be conducted to fully understand the roles of each of the estrogen receptors, there may be interesting implications of this work. For example, could a pharmacological agent that manipulates estrogen receptors improve mechanosensitivity and diminish the negative effects of estrogen deficiency on bone? In conjunction with an appropriate exercise program, could manipulation of estrogen receptors increase the effectiveness of exercise and improve bone mass? Novel treatments for postmenopausal osteoporosis and age-related bone loss may capitalize on the role of estrogen receptors in bone’s adaption to mechanical loading.
Oxidative Stress: New Mechanism for Aging-Related Bone Loss?
Oxidative stress has been a leading theory of aging for decades [47], but only very recently has it been advanced as a mechanism for age-related bone loss [48]. The primary mechanism leading to oxidative stress is the formation of reactive oxygen species (ROS) by electrons escaping from the electron transport chain during aerobic metabolism which then join with oxygen to form superoxide (O2 •), hydrogen peroxide (H2O2), and the hydroxyl radical (OH•) [48]. ROS cause damage to proteins, lipids, and DNA, initiating cell death and also trigger important signaling pathways, such as the p66shc pathway initiating cell apoptosis.
For several decades, it has been known that ROS in bone tissue are involved in the formation and activation of osteoclasts, thereby increasing bone resorption [49]. This led to an interesting hypothesis that oxidative stress could enhance, or even provide an alternate mechanism for, bone loss associated with menopause and aging. In a large-scale study of postmenopausal women, that subset with a diagnosis of osteoporosis had a higher oxidative status and lower antioxidant status as assessed from serum markers. There was a significant negative correlation between a key indicator of oxidative stress in this population and bone mineral density in the lumbar and femoral neck region [50].
Recent research has shown that estrogen may play a key role in protecting bone tissue against the damage caused by ROS. Estrogens have been identified as antioxidants in many tissues, effectively functioning as radical scavengers and suppressing peroxidation reactions [51]. Lean et al. [52] hypothesized that this antioxidant function of estrogen might be an important mechanism for its well-known salutary effect on bone health. Glutathione and thioredoxin, two major oxidative defense enzymes, are much reduced in rodent bone marrow after ovariectomy. A single dose of 17β-estradiol rapidly normalizes these antioxidant enzyme levels. More convincingly, administration of exogenous antioxidants, N-acetyl cysteine (NAC) and ascorbate, prevents bone loss in OVX mice (see Fig. 2.4) [52]. To further validate the effect of oxidative stress on bone, administration of l-buthionine-(S,R)-sulfoximine, an inhibitor of glutathione, resulted in similar loss of bone as observed in OVX animals. These data show, first of all, that ROS can lead to bone loss and, secondly, estrogens help to prevent bone loss by increasing intracellular oxidative defense mechanisms. The loss of estrogens at menopause could lead to a substantial decrease in the ability of the bone cells to defend against ROS and thereby contribute to increased osteoclastic activity and bone loss. Hence, the loss of estrogens in women at menopause may amplify the oxidative stress accounting for aging-related bone loss.
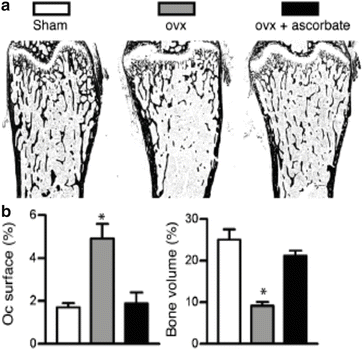
Fig. 2.4
Antioxidant treatment can reverse the impact of estrogen deficiency on cancellous bone mass and osteoclast surfaces. (a) Photomicrographs of distal femurs of mice subjected to sham ovariectomy (white bar), ovariectomy (gray bar), and ovariectomy with ascorbate treatment (black bar). Mineralized bone is in black. (b) Bone volume normalized to tissue volume (%BV/TV) and (b) Percent osteoclast surface (Oc.surface %) and cancellous bone volume normalized to tissue volume (%BV/TV) in the three groups. Adapted from Lean et al. [52]
Besides antioxidant enzymes (glutathione, thioredoxin, and superoxide dismutase), there are other pathways that lead to protection against ROS. The FoxO genes, so named for their unique structure with a special winged DNA helix known as a Forkhead box, are upregulated by ROS. Once FoxOs are activated and translocated into the nucleus of the cell, they lead to the transcription of free radical scavenging enzymes (MnSOD, catalase) and DNA repair genes (such as Gadd45). The FoxOs are also able to induce apoptosis in cells damaged by ROS [48, 53]. Mice null for FoxOs exhibit increased oxidative stress and a loss of both cortical and cancellous bone due to deficient bone formation [54]. If FoxO1 is conditionally deleted in osteoblasts, osteoblast proliferation and bone formation are significantly impaired. Administration of the antioxidant NAC normalizes osteoblast number, bone formation rate, and bone volume in mice lacking FoxO1 [55] and rates of osteoblast apoptosis in mice with osteoblast-specific deletions of FoxO1, O3, and O4 [56]. These studies further highlight the effects of oxidative stress on bone loss and the importance of defense mechanisms against an increase in ROS. Also of importance, exogenous antioxidant compounds are able to counteract the deleterious effects of the deletion of oxidative defense genes.
As Lean et al. hypothesized, the antioxidant abilities of estrogens may be critical to bone health due to its capacity to defend against oxidative stress [52]. Further research has shown that loss of estrogens and androgens accelerate the effects of aging by decreasing defenses against oxidative stress [48]. The loss of estrogen results in increased lipid peroxidation and hydrogen peroxide, and a decrease in oxidative defense enzymes (SOD, glutathione peroxidase, and glutathione S transferase) in femoral bone tissue of OVX mice, resulting in increased oxidative stress [57]. Almeida et al. compared normally aging mice (up to 31 months of age) of both genders with mice that underwent gonadectomy at 5 months of age [58]. Similar changes in ROS, glutathione reductase, and phosphorylation of p53 and p66shc were observed within 6 weeks of gonadectomy as observed over 31 months of aging. The administration of NAC protected against all measures of oxidative stress damage and was as effective at preserving spinal BMD as administration of either estrogen or testosterone. This exogenous antioxidant also mitigated the increase in osteocyte and osteoblast apoptosis caused by gonadectomy and maintained cancellous bone mass [58]. Later studies by the same laboratory confirm that antioxidants are as effective at maintaining bone integrity as sex steroid replacement [48, 54]. Taken together, these studies demonstrate the importance of oxidative defense mechanisms in bone tissue and demonstrate the powerful antioxidant effect of sex steroids.
Other potentially important mechanisms for estrogen’s antioxidant function relate to osteoblast and osteocyte apoptosis and to immune cell function. (Osteocytes are those cells embedded inside bone that are essential to signaling targeted remodeling of damaged bone, as well as sensing the mechanical loads placed on bone.) Pretreatment of MLO-Y4 osteocyte cells with either 17β-estradiol or selective estrogen receptor modulators (SERMs) prevents osteocyte apoptosis [59]. This suggests that estrogen deficiency may lead to a loss of osteocytes due to an inability to protect against ROS-induced damage. Oxidative stress also impacts osteoblast apoptosis. Aging or loss of androgens or estrogens leads to activation of nuclear factor-κB (NF-κB), and phosphorylation of p66shc, which increases the production of ROS and stimulates apoptosis. Administration of 17β-estradiol effectively inhibits ROS-stimulated activation of NF-κB and p66shc by acting on PKCβ, thereby attenuating osteoblast apoptosis [54]. OVX mice also exhibit an increase in ROS in bone marrow, which enhances the activity of bone marrow dendritic cells that activate T cells, leading to a signaling cascade resulting in bone loss. Bone loss is effectively prevented by supplying either an antioxidant or an inhibitor of the CD80/CD28 pathway, which is involved in T cell activation [60]. Taken together, these studies demonstrate that the loss of estrogen leads to a decreased ability to defend against ROS acting on key bone and immune cells important for maintaining bone integrity.
The evidence that oxidative stress leads to bone loss and the effect antioxidants exert on reversing this damage could lead to novel concepts for treating bone disorders like osteoporosis. Could a diet high in antioxidants help prevent the increase in ROS seen with aging? Could administration of antioxidants prevent the sharp decline in oxidative defense seen with postmenopausal osteoporosis and, therefore, preserve bone mass or attenuate its decline? In a cross-sectional study of postmenopausal women, Rao et al. [61] demonstrated a strong inverse correlation between serum levels of the antioxidant lycopene (found in tomatoes, red bell peppers, watermelon, and other red fruits and vegetables) with N-telopeptides (NTx), a well-accepted serum marker of bone resorption. There is also strong epidemiological evidence for the beneficial impact of flavonoids, polyphenolic compounds found in many plant foods that have anti-inflammatory and antioxidant properties, on bone health [62]. However, intervention trials provide less definitive conclusions to date.
Related posts:
 The Role of Estrogens in the Regulation of Peripheral Glucose Dynamics
The Role of Estrogens in the Regulation of Peripheral Glucose Dynamics
 Influence of Ovarian Hormones on Skeletal Muscle Contractility
Influence of Ovarian Hormones on Skeletal Muscle Contractility
 Transitions Across a Lifetime: Unique Cardiovascular Physiology of Women and Relationship to Cardiovascular Disease Risk
Transitions Across a Lifetime: Unique Cardiovascular Physiology of Women and Relationship to Cardiovascular Disease Risk
 Metabolic Health in the Aging Female: Human Perspective
Metabolic Health in the Aging Female: Human Perspective
 Estrogen Effects on Skeletal Muscle
Estrogen Effects on Skeletal Muscle
 The Impact of Estrogen Receptor α Expression in the Pathogenesis of the Metabolic Syndrome
The Impact of Estrogen Receptor α Expression in the Pathogenesis of the Metabolic Syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


