Chapter Outline
HEMOGLOBIN STRUCTURE AND FUNCTION
Introduction to Hemoglobin Variants
Identification of Hemoglobin Variants
Clinical Manifestations of Hemoglobin Variants Reflect Their Physiological Effects
Ontogeny of Globin Gene Expression Influences the Clinical Presentation of Hemoglobin Variants
Clinical Laboratory Tests to Identify Hemoglobin Variants
Clinical Management of Patients with Hemoglobin Variants
Studies of hemoglobin (Hb) structure, function, and expression over more than 50 years have produced many paradigms in biology and medicine. The first part of this chapter describes the structural and biochemical properties of Hb, emphasizing those that underlie its physiological roles. The conserved globin protein fold with the heme prosthetic group confers the basic function of reversible oxygen (O 2 ) binding, which is common to myoglobin (Mb), Hb, and numerous other recently discovered proteins such as neuroglobin and cytoglobin. The tetrameric quaternary structure of Hb allows for cooperative O 2 binding to achieve efficient loading of O 2 in lung capillaries at high partial pressure of O 2 (P O2 ≈100 mm Hg), and effective O 2 delivery to peripheral tissues at low P O2 (≈40 mm Hg). Several small molecules, including 2,3-diphosphoglycerate (2,3-DPG), chloride anion (Cl − ), and carbon dioxide (CO 2 )/bicarbonate dynamically regulate O 2 binding to Hb in response to varying physiological conditions. Hb also facilitates transport of CO 2 in the form of bicarbonate anion and participates in oxidation reactions, both beneficial and deleterious. The second part of this chapter reviews the biochemistry and pathophysiology of abnormal Hbs that give rise to human diseases. Globin gene mutations that alter the properties of Hbs are common, affecting about 7% of the world’s population, and present a significant global health problem.
This chapter reviews Hb structure, biochemistry, and mutations that produce abnormal globin proteins. These “Hb variants” are usually caused by single amino acid substitutions, but also by amino acid insertions and/or deletions, antitermination mutations and altered posttranslational processing. The most common pathological Hb variants, HbS and HbC are reviewed in Chapter 20 . Other variants, discussed in the second part of this chapter, number more than a thousand and are rare individually but common collectively. Globin gene mutations that impair protein subunit production (i.e., thalassemias) are discussed in Chapter 21 . Several excellent references review the concepts discussed in this chapter.
Hemoglobin Structure and Function
The study of Hb and related proteins has contributed immensely to the understanding of protein function in general. In 1957, Kendrew and colleagues used X-ray diffraction to solve the structure of Mb at 6-Å resolution and a few years later refined this structure to 2 Å, making it possible to build models at near atomic resolution. These remarkable achievements showed for the first time what a protein actually “looks like.” Almost simultaneously the structure of Hb was determined at 5.5-Å resolution by Perutz and colleagues. Since then numerous biophysical studies have further refined the structures of normal and mutant Hbs to resolutions approaching 1 Å.
HbA, the major adult form of Hb, is a tetramer of two α polypeptide chains and two β polypeptide chains (α 2 β 2 ), each bound to a heme prosthetic group ( Box 19-1 ). The α and β globins are believed to have arisen from the duplication of a common ancestral gene approximately 450 million years ago, around the time that the lineages of cartilaginous fish and bony fish diverged. Subsequent gene duplications produced additional globin genes that are expressed at different times during ontogeny. During early human embryogenesis, the α-like (ζ) and β-like (ε and γ) chains produce embryonic Hbs (ζ 2 ε 2 , ζ 2 γ 2 , and α 2 ε 2 ). During midgestation, fetal Hb (HbF; α 2 γ 2 ) is predominantly expressed. From birth until around postnatal week 12, β-globin expression gradually increases and γ globin decreases as HbA replaces HbF. Another β-like globin (δ) is also expressed postnatally, and normal hemolysates contain approximately 2% HbA 2 (α 2 δ 2 ). The properties of embryonic and fetal globins are tuned to the physiological conditions experienced in utero, particularly low O 2 tension, although the functions of all human Hb isoforms can be understood as variations on the mechanism of adult HbA. The onset of symptoms caused by deleterious globin gene mutations varies according to the developmental expression pattern of each particular gene, as discussed later in this chapter.
- •
The globin fold is an evolutionary conserved structure made up of seven or eight α helices folded into a globular shape.
- •
HbA is a tetramer made up of two α- and two β-globin subunits, each linked to a heme moiety that binds O 2 reversibly via a central iron molecule. Only reduced iron (Fe[II]) can bind O 2 .
- •
Hb can bind other diatomic ligands via heme iron, including CO and NO.
- •
The α1β1 (and α2β2) dimer is the basic structural unit of hemoglobin.
- •
Various α- and β-like globins are expressed during human ontogeny. These include:
- •
Embryonic: Gower 1 (ζ 2 ε 2 ), Gower 2 (α 2 ε 2 ), Hb Portland I (ζ 2 γ 2 ), Hb Portland II (ζ 2 β 2 )
- •
Fetal: HbF (α 2 γ 2 )
- •
Adult: HbA (α 2 β 2 ), ~95%; HbA 2 (α 2 δ 2 ), 1.5%-3.5%; HbF (α 2 γ 2 ), 1%-2%.
- •
CO, Carbon monoxide; Hb, hemoglobin; NO, nitric oxide; O 2 , oxygen.
Hemoglobin Primary Structure
Primary structure refers to the linear sequence of amino acids within a protein. The primary structures of the α-like and β-like globin chains are shown in Table 19-1 . Translation of the polypeptide chains on the ribosome is initiated with methionine, which is subsequently removed by methionine aminopeptidase before assembly of mature Hb tetramer. The α (141 amino acid residues) and β (146 amino acid residues) chains share only 41% amino acid sequence identity, yet they adopt very similar three-dimensional (3D; tertiary) structures. Numerous naturally occurring mutations alter the primary sequence of globin proteins (see the second part of this chapter).
| Helix | α | ζ | Helix | β | δ | γ | ε |
|---|---|---|---|---|---|---|---|
| NA1 | 1 Val | Ser | NA1 NA2 | 1 Val 2 His | Val His | Gly His | Val His |
| NA2 | 2 Leu | Leu | NA3 | 3 Leu | Leu | Phe | Phe |
| A1 | 3 Ser | Thr | A1 | 4 Thr | Thr | Thr | Thr |
| A2 | 4 Pro | Lys | A2 | 5 Pro | Pro | Glu | Ala |
| A3 | 5 Ala | Thr | A3 | 6 Glu | Glu | Glu | Glu |
| A4 | 6 Asp | Glu | A4 | 7 Glu | Glu | Asp | Glu |
| A5 | 7 Lys | Arg | A5 | 8 Lys | Lys | Lys | Lys |
| A6 | 8 Thr | Thr | A6 | 9 Ser | Thr | Ala | Ala |
| A7 | 9 Asn | Ile | A7 | 10 Ala | Ala | Thr | Ala |
| A8 | 10 Val | Ile | A8 | 11 Val | Val | Ile | Val |
| A9 | 11 Lys | Val | A9 | 12 Thr | Asn | Thr | Thr |
| A10 | 12 Ala | Ser | A10 | 13 Ala | Ala | Ser | Ser |
| A11 | 13 Ala | Met | A11 | 14 Leu | Leu | Leu | Leu |
| A12 | 14 Trp | Trp | A12 | 15 Trp | Trp | Trp | Trp |
| A13 | 15 Gly | Ala | A13 | 16 Gly | Gly | Gly | Ser |
| A14 | 16 Lys | Lys | A14 | 17 Lys | Lys | Lys | Lys |
| A15 | 17 Val | Ile | A15 | 18 Val | Val | Val | Met |
| A16 | 18 Gly | Ser | |||||
| AB1 | 19 Ala | Thr | |||||
| B1 | 20 His | Gln | B1 | 19 Asn | Asn | Asn | Asn |
| B2 | 21 Ala | Ala | B2 | 20 Val | Val | Val | Val |
| B3 | 22 Gly | Asp | B3 | 21 Asp | Asp | Glu | Glu |
| B4 | 23 Glu | Thr | B4 | 22 Glu | Ala | Asp | Glu |
| B5 | 24 Tyr | Ile | B5 | 23 Val | Val | Ala | Ala |
| B6 | 25 Gly | Gly | B6 | 24 Gly | Gly | Gly | Gly |
| B7 | 26 Ala | Thr | B7 | 25 Gly | Gly | Gly | Gly |
| B8 | 27 Glu | Glu | B8 | 26 Glu | Glu | Glu | Glu |
| B9 | 28 Ala | Thr | B9 | 27 Ala | Ala | Thr | Ala |
| B10 | 29 Leu | Leu | B10 | 28 Leu | Leu | Leu | Leu |
| B11 | 30 Glu | Glu | B11 | 29 Gly | Gly | Gly | Gly |
| B12 | 31 Arg | Arg | B12 | 30 Arg | Arg | Arg | Arg |
| B13 | 32 Met | Leu | B13 | 31 Leu | Leu | Leu | Leu |
| B14 | 33 Phe | Phe | B14 | 32 Leu | Leu | Leu | Leu |
| B15 | 34 Leu | Leu | B15 | 33 Val | Val | Val | Val |
| B16 | 35 Ser | Ser | B16 | 34 Val | Val | Val | Val |
| C1 | 36 Phe | His | C1 | 35 Tyr | Tyr | Tyr | Tyr |
| C2 | 37 Pro | Pro | C2 | 36 Pro | Pro | Pro | Pro |
| C3 | 38 Thr | Gln | C3 | 37 Trp | Trp | Trp | Trp |
| C4 | 39 Thr | Thr | C4 | 38 Thr | Thr | Thr | Thr |
| C5 | 40 Lys | Lys | C5 | 39 Gln | Gln | Gln | Gln |
| C6 | 41 Thr | Thr | C6 | 40 Arg | Arg | Arg | Arg |
| C7 | 42 Tyr | Tyr | C7 | 41 Phe | Phe | Phe | Phe |
| CE1 | 43 Phe | Phe | CD1 | 42 Phe | Phe | Phe | Phe |
| CE2 | 44 Pro | Pro | CD2 | 43 Glu | Glu | Asp | Asp |
| CE3 | 45 His | His | CD3 | 44 Ser | Ser | Ser | Ser |
| CE4 | 46 Phe | Phe | CD4 | 45 Phe | Phe | Phe | Phe |
| CD5 | 46 Gly | Gly | Gly | Gly | |||
| CE5 | 47 Asp | Asp | CD6 | 47 Asp | Asp | Asn | Asn |
| CE6 | 48 Leu | Leu | CD7 | 48 Leu | Leu | Leu | Leu |
| CE7 | 49 Ser | His | CD8 | 49 Ser | Ser | Ser | Ser |
| CE8 | 50 His | Pro | D1 | 50 Thr | Ser | Ser | Ser |
| D2 | 51 Pro | Pro | Ala | Pro | |||
| D3 | 52 Asp | Asp | Ser | Ser | |||
| D4 | 53 Ala | Ala | Ala | Ala | |||
| D5 | 54 Val | Val | Ile | Ile | |||
| D6 | 55 Met | Met | Met | Leu | |||
| CE9 | 51 Gly | Gly | D7 | 56 Gly | Gly | Gly | Gly |
| E1 | 52 Ser | Ser | E1 | 57 Asn | Asn | Asn | Asn |
| E2 | 53 Ala | Ala | E2 | 58 Pro | Pro | Pro | Pro |
| E3 | 54 Gln | Gln | E3 | 59 Lys | Lys | Lys | Lys |
| E4 | 55 Val | Leu | E4 | 60 Val | Val | Val | Val |
| E5 | 56 Lys | Arg | E5 | 61 Lys | Lys | Lys | Lys |
| E6 | 57 Gly | Ala | E6 | 62 Ala | Ala | Ala | Ala |
| E7 | 58 His | His | E7 | 63 His | His | His | His |
| E8 | 59 Gly | Gly | E8 | 64 Gly | Gly | Gly | Gly |
| E9 | 60 Lys | Ser | E9 | 65 Lys | Lys | Lys | Lys |
| E10 | 61 Lys | Lys | E10 | 66 Lys | Lys | Lys | Lys |
| E11 | 62 Val | Val | E11 | 67 Val | Val | Val | Val |
| E12 | 63 Ala | Val | E12 | 68 Leu | Leu | Leu | Leu |
| E13 | 64 Asp | Ser | E13 | 69 Gly | Gly | Thr | Thr |
| E14 | 65 Ala | Ala | E14 | 70 Ala | Ala | Ser | Ser |
| E15 | 66 Leu | Val | E15 | 71 Phe | Phe | Leu | Phe |
| E16 | 67 Thr | Gly | E16 | 72 Ser | Ser | Gly | Gly |
| E17 | 68 Asn | Asp | E17 | 73 Asp | Asp | Asp | Asp |
| E18 | 69 Ala | Ala | E18 | 74 Gly | Gly | Ala | Ala |
| E19 | 70 Val | Val | E19 | 75 Leu | Leu | Ile, Thr | Ile |
| E20 | 71 Ala | Lys | E20 | 76 Ala | Ala | Lys | Lys |
| EF1 | 72 His | Ser | EF1 | 77 His | His | His | Asn |
| EF2 | 73 Val | Ile | EF2 | 78 Leu | Leu | Leu | Met |
| EF3 | 74 Asp | Asp | EF3 | 79 Asp | Asp | Asp | Asp |
| EF4 | 75 Asp | Asp | EF4 | 80 Asn | Asn | Asp | Asn |
| EF5 | 76 Met | Ile | EF5 | 81 Leu | Leu | Leu | Leu |
| EF6 | 77 Pro | Gly | EF6 | 82 Lys | Lys | Lys | Lys |
| EF7 | 78 Asn | Gly | EF7 | 83 Gly | Gly | Gly | Pro |
| EF8 | 79 Ala | Ala | EF8 | 84 Thr | Thr | Thr | Ala |
| F1 | 80 Leu | Leu | F1 | 85 Phe | Phe | Phe | Phe |
| F2 | 81 Ser | Ser | F2 | 86 Ala | Ser | Ala | Ala |
| F3 | 82 Ala | Lys | F3 | 87 Thr | Gln | Gln | Lys |
| F4 | 83 Leu | Leu | F4 | 88 Leu | Leu | Leu | Leu |
| F5 | 84 Ser | Ser | F5 | 89 Ser | Ser | Ser | Ser |
| F6 | 85 Asp | Glu | F6 | 90 Glu | Glu | Glu | Glu |
| F7 | 86 Leu | Leu | F7 | 91 Leu | Leu | Leu | Leu |
| F8 | 87 His | His | F8 | 92 His | His | His | His |
| F9 | 88 Ala | Ala | F9 | 93 Cys | Cys | Cys | Cys |
| FG1 | 89 His | Tyr | FG1 | 94 Asp | Asp | Asp | Asp |
| FG2 | 90 Lys | Ile | FG2 | 95 Lys | Lys | Lys | Lys |
| FG3 | 91 Leu | Leu | FG3 | 96 Leu | Leu | Leu | Leu |
| FG4 | 92 Arg | Arg | FG4 | 97 His | His | His | His |
| FG5 | 93 Val | Val | FG5 | 98 Val | Val | Val | Val |
| G1 | 94 Asp | Asp | G1 | 99 Asp | Asp | Asp | Asp |
| G2 | 95 Pro | Pro | G2 | 100 Pro | Pro | Pro | Pro |
| G3 | 96 Val | Val | G3 | 101 Glu | Glu | Glu | Glu |
| G4 | 97 Asn | Asn | G4 | 102 Asn | Asn | Asn | Asn |
| G5 | 98 Phe | Phe | G5 | 103 Phe | Phe | Phe | Phe |
| G6 | 99 Lys | Lys | G6 | 104 Arg | Arg | Lys | Lys |
| G7 | 100 Leu | Leu | G7 | 105 Leu | Leu | Leu | Leu |
| G8 | 101 Leu | Leu | G8 | 106 Leu | Leu | Leu | Leu |
| G9 | 102 Ser | Ser | G9 | 107 Gly | Gly | Gly | Gly |
| G10 | 103 His | His | G10 | 108 Asn | Asn | Asn | Asn |
| G11 | 104 Cys | Cys | G11 | 109 Val | Val | Val | Val |
| G12 | 105 Leu | Leu | G12 | 110 Leu | Leu | Leu | Met |
| G13 | 106 Leu | Leu | G13 | 111 Val | Val | Val | Val |
| G14 | 107 Val | Val | G14 | 112 Cys | Cys | Thr | Ile |
| G15 | 108 Thr | Thr | G15 | 113 Val | Val | Val | Ile |
| G16 | 109 Leu | Leu | G16 | 114 Leu | Leu | Leu | Leu |
| G17 | 110 Ala | Ala | G17 | 115 Ala | Ala | Ala | Ala |
| G18 | 111 Ala | Ala | G18 | 116 His | Arg | Ile | Thr |
| G19 | 112 His | Arg | G19 | 117 His | Asn | His | His |
| GH1 | 113 Leu | Phe | GH1 | 118 Phe | Phe | Phe | Phe |
| GH2 | 114 Pro | Pro | GH2 | 119 Gly | Gly | Gly | Gly |
| GH3 | 115 Ala | Ala | GH3 | 120 Lys | Lys | Lys | Lys |
| GH4 | 116 Glu | Asp | GH4 | 121 Glu | Glu | Glu | Glu |
| GH5 | 117 Phe | Phe | GH5 | 122 Phe | Phe | Phe | Phe |
| H1 | 118 Thr | Thr | H1 | 123 Thr | Thr | Thr | Thr |
| H2 | 119 Pro | Ala | H2 | 124 Pro | Pro | Pro | Pro |
| H3 | 120 Ala | Glu | H3 | 125 Pro | Gln | Glu | Glu |
| H4 | 121 Val | Ala | H4 | 126 Val | Met | Val | Val |
| H5 | 122 His | His | H5 | 127 Gln | Gln | Gln | Gln |
| H6 | 123 Ala | Ala | H6 | 128 Ala | Ala | Ala | Ala |
| H7 | 124 Ser | Ala | H7 | 129 Ala | Ala | Ser | Ala |
| H8 | 125 Leu | Trp | H8 | 130 Tyr | Tyr | Trp | Trp |
| H9 | 126 Asp | Asp | H9 | 131 Gln | Gln | Gln | Gln |
| H10 | 127 Lys | Lys | H10 | 132 Lys | Lys | Lys | Lys |
| H11 | 128 Phe | Phe | H11 | 133 Val | Val | Met | Leu |
| H12 | 129 Leu | Leu | H12 | 134 Val | Val | Val | Val |
| H13 | 130 Ala | Ser | H13 | 135 Ala | Ala | Thr | Ser |
| H14 | 131 Ser | Val | H14 | 136 Gly | Gly | Gly, Ala | Ala |
| H15 | 132 Val | Val | H15 | 137 Val | Val | Val | Val |
| H16 | 133 Ser | Ser | H16 | 138 Ala | Ala | Ala | Ala |
| H17 | 134 Thr | Ser | H17 | 139 Asn | Asn | Ser | Ile |
| H18 | 135 Val | Val | H18 | 140 Ala | Ala | Ala | Ala |
| H19 | 136 Leu | Leu | H19 | 141 Leu | Leu | Leu | Leu |
| H20 | 137 Thr | Thr | H20 | 142 Ala | Ala | Ser | Ala |
| H21 | 138 Ser | Glu | H21 | 143 His | His | Ser | His |
| HC1 | 139 Lys | Lys | HC1 | 144 Lys | Lys | Arg | Lys |
| HC2 | 140 Tyr | Tyr | HC2 | 145 Tyr | Tyr | Tyr | Tyr |
| HC3 | 141 Arg | Arg | HC3 | 146 His | His | His | His |
* The α and α-related (ζ) subunits are shown at the left. The β and β-related (δ, γ, and ε) chains are at the right. The amino acids’ relationship to the eight globin helices (A to H) is also shown. Thus A16 is the sixteenth amino acid in the A helix. Interhelical elbows are named for the two adjacent helices (e.g., AB1 is the first amino acid between helices A and B). The N- and C-terminal residues are labeled NA and HC, respectively. The residues are aligned to maximize the homology between subunits, which causes some gaps.
Hemoglobin Secondary Structure
Secondary structure refers to the organization of primary structure into sections of regular polypeptide backbone geometry. Hb chains are comprised primarily of α-helical secondary structure joined by turns or short sections with irregular polypeptide backbone conformation. The geometry of regular secondary structure is dictated by steric constraints (clashes between atoms in neighboring residues) and maximization of hydrogen bonding between amide and carbonyl groups in the backbone. Although hydrogen bonds are relatively weak individually, their repeating pattern in α helices has a cooperative effect to make the structure comparatively rigid. Through this property, α helices transmit forces in proteins and protein complexes to alter their structure and modulate function, as occurs for Hb. Most globin chains contain seven or eight α helices labeled A through H (from amino to carboxyl terminus), with the intervening links referred to as corners or loops. The position of each residue is described with reference to these structural elements. For example, the heme-coordinating histidine occurs at position F8 (residue 8 in helix F) in all globins (see Table 19-1 ).
Hemoglobin Tertiary Structure
Tertiary structure refers to the 3D structure of a polypeptide that is formed by packing secondary structural elements together. In general, hydrophobic side chains occupy the interior of a protein or protein domain, and polar and charged residues are arranged on the exterior ( Fig. 19-1, A and B ). Thus mutations converting a nonpolar interior amino acid to a polar one commonly produce an unstable protein (see the second part of this chapter). The α-helical segments of globin chains fold together into a globular shape with a cavity that is occupied by the O 2 -binding heme prosthetic group (see Fig. 19-1, B -D ). The basic tertiary structure, called the globin fold, occurs in a large, ancient protein family with members expressed in animals, plants, and bacteria. Figure 19-2 shows the tertiary structures of α and β globin with conserved α-helical arrangements and amino acid residues that mediate Hb function. Importantly, these structures are not static or rigid. Rather the tertiary structures of α and β globins (and the quaternary structure of the tetramer) change according to binding of O 2 or other ligands and also through interactions with allosteric effectors. These dynamic structural properties underlie the physiological function of Hb as a gas transporter and the pathologic features of many Hb variants.
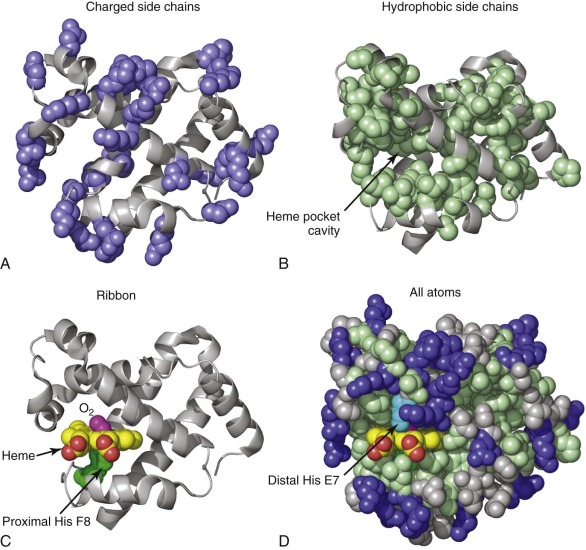
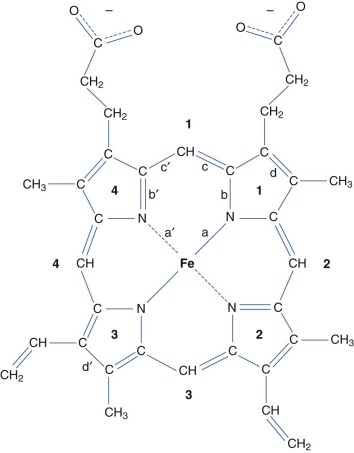
The Prosthetic Heme Group
Each α and β chain binds one heme b group ( Fig. 19-3 ), which forms the binding site for O 2 (see Fig. 19-1, C ). Heme is an amphipathic molecule that is highly insoluble in water, so it must be bound to protein carriers or protein active sites under physiologic conditions. Biosynthesis of protoporphyrin IX occurs through eight well-characterized steps that take place either in the cytosol or the intermembrane space of mitochondria. The final step, insertion of an iron ion, is catalyzed by the enzyme ferrochelatase.
In the folded α and β chains, heme is bound into a deep pocket that is lined with hydrophobic residues (see Fig. 19-1, B and D ). Interactions between the protein and heme are critical for stabilizing the native globin fold. Thus mutations that destabilize heme–protein interactions lead to increased globin precipitation and the release of free heme, which has pathologic consequences (see the second part of this chapter). A coordinate covalent bond between the heme iron and the Nε atom of a histidine residue in the co-factor binding pocket, termed the proximal histidine (F8) , is of primary importance for globin structure and O 2 binding function (see Fig. 19-1, C and Residues of the Heme Pocket). A sixth iron–coordination site on the opposite (distal) side of the planar heme is available for reversible O 2 binding (see Fig. 19-1, C ). In addition to direct iron coordination to the proximal histidine, the following heme–globin interactions are important for maintaining the Hb tertiary structure: 1) hydrophobic interactions between nonpolar residues in the pocket and the porphyrin ring; 2) hydrogen bonding between residues in the distal heme pocket and diatomic ligands; and 3) electrostatic interactions between the heme propionates and amino acids on the surface of the protein. An asymmetric distribution of methyl and vinyl side groups on the heme ring defines a stereospecific orientation of heme binding. Heme iron can exist in a range of oxidation states in vivo, most commonly as Fe(II) or Fe(III), but the ferryl Fe(IV) is also biologically relevant (see Production of Ferryl Heme and Hemoglobin Protein Radicals ). Importantly, only Fe(II) (ferrous) heme can bind O 2 . The distal histidine (E7) of globin chains typically stabilizes bound O 2 , but under certain circumstances can also bind the sixth coordinate position of heme iron to form structures termed hemichromes or hemochromes when the iron is oxidized or reduced, respectively.
Hemoglobin Quaternary Structure
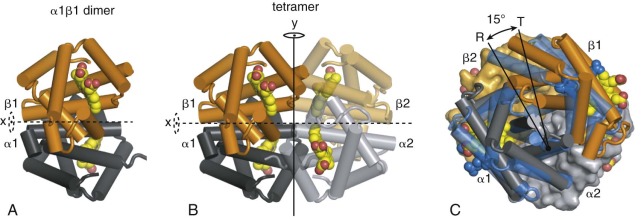
Quaternary structure refers to the packing of protein subunits into a multimeric complex. During Hb assembly, α and β monomers join to form a very high-affinity αβ dimer (association equilibrium constant [ K A ] >10 10 M −1 ) ( Fig. 19-4, A ). Two identical α1β1 and α2β2 dimers interact to form a tetramer, in which the constituent dimers are related by a two-fold rotational (C2) symmetry (see Fig. 19-4, B ). This interaction between dimers is coupled to O 2 ligand binding, with deoxygenated dimers having a higher affinity for self-association to form tetramers ( K A ≈4 × 10 10 M −1 ) than fully oxygenated dimers ( K A ≈10 6 M −1 ). The major dimer–dimer contacts involve nonidentical chains at the α1β2 and α2β1 interfaces. Changes in the interactions between α1β1 and α2β2 dimers are central to the mechanism of cooperative O 2 binding (see Fig. 19-4, C ).
Free globin subunits, particularly the apo forms lacking heme, are unstable and toxic as monomers and must be assembled rapidly and efficiently into Hb tetramers during erythropoiesis. The order of globin subunit assembly into mature Hb tetramers is unknown, but probably involves parallel pathways. For example, heme may be incorporated into apo α or β globins before or after their assembly into αβ dimers. In mammals, nascent apo or met α globins may be bound and stabilized by the molecular chaperone alpha Hb stabilizing protein (AHSP) during the early steps of Hb assembly in vivo.
Dioxygen Binding
The Oxygen Carrying Capacity of Hemoglobin
The solubility of O 2 in water (≈0.3 mM at 20° C and 1 atm) is insufficient to achieve full tissue oxygenation in most animals. Consequently animals have evolved to use heme or other respiratory pigments to transport O 2 . However, O 2 rapidly oxidizes free heme from the Fe(II) to Fe(III) state, which cannot bind O 2 (see Redox Reactions of Hemoglobin ). To overcome this potential limitation, globin proteins protect the heme group from oxidation and fine tune ligand binding to allow for reversible O 2 uptake and delivery. Hb carries O 2 from the lungs to peripheral tissue capillaries and CO 2 in the reverse direction. The concentration of Hb tetramers in red blood cells (RBCs) is typically about 5.2 mM (equivalent to ≈21 mM heme), and RBCs typically account for 45% of blood volume, giving a theoretical O 2 concentration of approximately 9 mM. O 2 is 21% by volume in air, which corresponds to roughly 8.75 mM in the gas phase at 1 atm and 20° C (1 mol gas under these conditions occupies 24 L by the ideal gas law). Hence Hb raises the O 2 concentration in blood to the same level as in air.
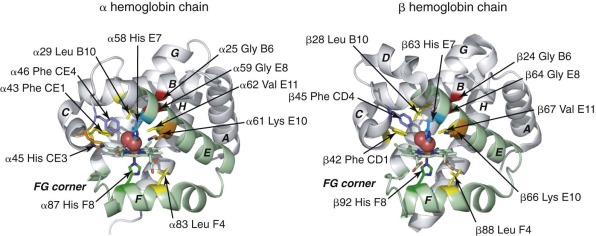
Residues of the Heme Pocket
Conserved, functionally important residues in the heme pockets of α and β globin are shown in Figure 19-2 . The proximal His-F8 stabilizes an axial iron ligand, which is essential for establishing the octahedral metal coordination geometry that is necessary for O 2 binding. Electrostatic interactions between bound O 2 and the distal His-E7 are of primary importance for ligand selectivity ( Fig. 19-5 ) and inhibiting heme oxidation (autoxidation). In both α and β globin, hydrogen bonding between His-E7 and O 2 has been identified through high resolution (1.25 Å) x-ray crystal structures, nuclear magnetic resonance, electron paramagnetic resonance (EPR), and site-directed mutagenesis. This interaction contributes around 1.9 kcal mol −1 to O 2 binding affinity. The importance of this interaction is illustrated by an approximate thousandfold increase in the O 2 dissociation rate from Mb when the distal His is replaced with a nonpolar amino acid. A number of nonpolar side chains in the proximal heme pocket make important steric interactions with bound O 2 (see Fig. 19-2 , yellow ). In particular, αPhe CE1/βPhe CD1, which is highly conserved in all globins, stabilizes the heme group and helps orient bound O 2 and His-E7 for efficient hydrogen bonding. βPhe-CD4 (αPhe-CE4) also plays a role in positioning of the distal His side chain to enable O 2 binding (see Fig. 19-2 ). In the proximal heme pocket, Leu F4 prevents water from entering and disrupting the His F8–iron bond.
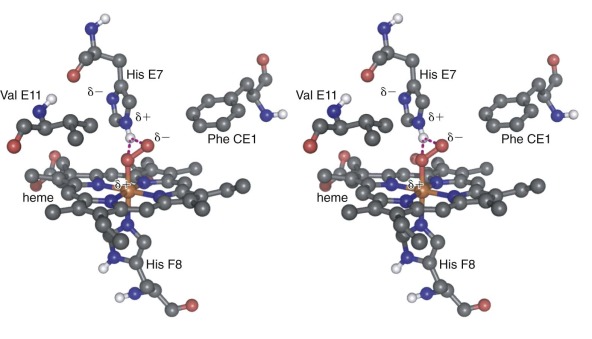
The heme pocket of globins is relatively enclosed (see Fig 19-1, D ), and multiple routes of O 2 entry have proposed, based on binding studies of xenon gas into nonpolar cavities of Mb and Hb. X-ray crystallographic and kinetic analyses of Mb and Hb indicate that O 2 enters via a channel caused by outward rotation of the distal His-E7 (see Figs. 19-1, D , and 19-2 , cyan ), which allows direct entry into the pocket rather than migration through other cavities in the protein.
In addition to O 2 binding, ferrous Fe(II) Hb also binds carbon monoxide (CO) and nitric oxide (NO) (see Nitric Oxide–Hemoglobin Reactions ). Studies with model porphyrin systems show that the Fe–CO bond is intrinsically stronger than the Fe–O 2 bond by a factor of 2 × 10 4 . If this ratio of affinities were true in the context of an Hb tetramer, then humans would be rapidly poisoned by endogenously produced CO, which is generated by heme catabolism, neuronal function, and cell signaling in the vasculature. High-level environmental CO exposure can also occur from cigarette smoke and internal combustion engines. Thankfully the relative affinity of CO and O 2 is reduced from 2 × 10 4 in free porphyrins to about 220 in Hb, largely because of the different electrostatic properties of the Fe–CO and Fe–O 2 complexes. The Fe–O 2 complex is highly polar and makes strong hydrogen bonding interactions with the distal His E7 (see Fig. 19-5 ). In contrast, the nonpolar Fe–CO complex interacts only weakly with the distal His.
Cooperative Oxygen Binding
Binding of O 2 to Mb can be described by a simple one–binding-site model that yields a hyperbolic curve ( Box 19-2 and Fig. 19-6 ):
Y = P O 2 / ( P 50 + P O 2 )
- •
Cooperative O 2 binding results when the binding of O 2 to one site in Hb increases the O 2 affinity of the remaining sites. Similarly, release of O 2 from one site increases the probability that O 2 will be released from the remaining unbound sites. The overall effect enhances the amount of O 2 that can be bound and released over the narrow range of P O2 values that characterize arterial and venous blood (see Fig. 19-6 ).
- •
Hb tetramers can bind four O 2 molecules (one per subunit). Binding of several O 2 molecules to deoxy-Hb causes a switch to a high affinity conformation. The high–O 2 affinity Hb quaternary structure is referred to as relaxed (R), and the low-affinity Hb structure is referred to as tense (T).
- •
O 2 binding to the heme iron of one Hb subunit alters quaternary structure at the α1β2 and α2β1 interfaces of the tetramer, leading to structural changes in the heme pockets of unliganded Hb subunits to enhance their O 2 affinity. As a result, some mutations that affect the α1β2 and α2β1 interfaces alter the O 2 equilibrium curve, causing right or left shifts and different amounts of O 2 transport in vivo.
deoxy-Hb, Deoxygenated hemoglobin; Hb, hemoglobin; O 2 , oxygen; P O2 , partial pressure of oxygen.
In this equation, Y is the fractional O 2 saturation, P O2 is the partial pressure of O 2 , and P 50 is the value of P O2 at which 50% of the O 2 -binding sites are occupied. For the simple one–binding-site equilibrium, the dissociation constant, K d , is equal to P 50 . In contrast, the O 2 equilibrium curve for Hb is sigmoidal (see Fig. 19-6 ). The initial portion of the curve has a small slope, indicating that fully deoxygenated Hb (deoxy-Hb) has a relatively low affinity for O 2 . As O 2 loading proceeds, the slope of the curve steepens, reflecting increasing Hb affinity for O 2 . Ultimately, the slope levels off again as Hb approaches O 2 saturation. The shape of this curve indicates that O 2 binding to one or more sites causes an increase in the O 2 affinity of the remaining sites. This property, called positive cooperativity, is essential for Hb function.
Conversely, deoxygenation at one or more Hb sites decreases O 2 affinity for the remaining sites, favoring its release in peripheral tissues where O 2 concentration is relatively low. In this way, approximately 1.7 times more O 2 can be delivered to the tissues than would be possible with a hypothetical noncooperative O 2 binding protein with the same intrinsic O 2 affinity. An empirical relationship that described cooperative O 2 binding activity was proposed by A.V. Hill in 1910.
Y = ( P O 2 ) n / ( P 50 ) n + ( P O 2 ) n
Although the Hill equation reproduces the observed O 2 binding of Hb reasonably well, it implies simultaneous binding of multiple O 2 ligands. G.S. Adair determined that Hb contained four binding sites for O 2 and formulated a sequential binding model in which each O 2 binds with progressively higher affinity to each of the four sites. Adair’s equation gives an accurate fit to the experimental O 2 equilibrium curve and uses a general and physically plausible binding model. However, the generality of the model means that it does not provide any mechanistic insight into Hb cooperativity. In fact, cooperative binding implies some communication between the heme molecules in each globin chain. Before the structure of Hb (or any protein) was determined, Linus Pauling formulated a model based on a direct chemical interaction between heme groups and showed that O 2 binding characteristics could be reproduced, assuming that subunits of Hb were symmetrically arranged such that O 2 at one heme could influence binding at two adjacent sites. Given the distant spacing between the heme groups, communication must be mediated by the Hb protein matrix, with binding at one site sterically affecting O 2 binding to a heme group at another (“allo-”) site on an adjacent subunit.
Allostery and the Perutz–Monod, Wyman, Changeux Model of Hemoglobin Function
The term allosteric was first coined in 1961 by Monod and Jacob to described how regulatory ligands could influence the activity of enzymes by binding to distinct sites that did not overlap with the substrate binding site. Allostery, an extremely important concept in biochemistry, has been described as “the second secret of life,” second only to the genetic code. The Monod, Wyman, Changeux (MWC) model, developed in the early 1960s, still forms a theoretical basis for understanding cooperative O 2 binding to Hb. The theory itself was influenced by the structural view of Hb provided by Perutz. A key finding from Perutz and colleagues was the elucidation of the human deoxy-Hb structure at 5.5 Å, which provided an opportunity for comparison with the earlier structure of horse oxygenated Hb (HbO 2 ), also at 5.5 Å. At this resolution no difference in the structures of the individual subunits could be clearly identified. However, there was a clear difference in the quaternary structure. Perutz and colleagues noted that, “the two β chains in human reduced (deoxy-)Hb have moved apart, increasing the distance between their two haem groups 7 Å compared with that in horse oxyhaemoglobin.” This quaternary structural rearrangement of Hb in response to O 2 binding is at the heart of the MWC model.
The MWC model is shown in Figure 19-7, A . Its basic principle is that quaternary structural changes in Hb regulate the O 2 -binding affinity of the subunit chains. The MWC theory describes Hb as a symmetrical oligomer that can adopt two distinct quaternary structures, termed the tense (T) and relaxed (R) states or conformations. The T and R states are in equilibrium, and in the absence of O 2 , the T state is much more stable than the R state (see Fig. 19-7, A, top row ). The equilibrium between the T and R states of unliganded Hb is defined by the MWC parameter, L = [T 0 ]/[R 0 ], where [T 0 ] and [R 0 ] are the molar concentrations of the zero-liganded T and R states. In the T state, all the subunits have a tertiary conformation with low–O 2 affinity (small association equilibrium constant K T or high P 50 ), whereas in the R state, all subunits have a tertiary conformation with high–O 2 affinity (large association equilibrium constant K R or low P 50 ). Note that O 2 binding to the individual T (or R) state tetramers does not increase the affinity of the T (or R) state for binding additional O 2 molecules, so there is no cooperativity within the T (or R) quaternary state. However, because O 2 preferentially binds to empty sites in the R quaternary state rather than the T state ( K R > K T ), the T→R equilibrium must shift towards R with the addition of O 2 , according to the Le Châtelier principle. The basis for cooperativity is that the T→R quaternary switch involves a concerted switch of all subunits from the low–O 2 affinity tertiary structure to the high–O 2 affinity structure ( right-to-left shifts in Fig. 19-7, A ). The level of cooperativity is determined by the parameters L and c = K T / K R . The MWC model provides a good fit to experimental equilibrium O 2 -binding data, yielding typical parameters of L ≈ 5×10 6 and c ≈ 1.5 ×10 −3 and 1/ K T ≈ 80 torr for pH 7.4, 0.15 M NaCl, 21.5° C. These parameters are significantly modulated by pH, salt, and organic phosphate concentration (see Heterotropic Effectors of Oxygen Binding to Hemoglobin ). The parameter c reveals that the quaternary T→R switch corresponds to a 500- to 1000-fold increase in O 2 affinity. The MWC model neglects differences between α and β affinities, but these are likely to differ by less that twofold. Many excellent reviews of the MWC model with expanded and refined models are available.
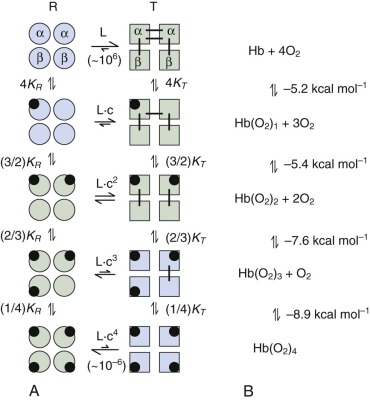
In order to move from the unliganded T 0 state to the fully O 2 -liganded R 4 state, the bonding and nonbonding interactions that stabilize the T 0 relative to R 0 quaternary structures (see lines joining T-state subunits in Fig. 19-7, A ) must be broken using free energy provided by O 2 binding. Consequently the free energy for binding of each successive O 2 becomes more favorable (see Fig. 19-7, B ). In the unconstrained R state, each binding event would yield −8.35 kcal mol −1 . The difference between the free energy for binding four O 2 molecules in HbA (−27.1 kcal mol −1 ) compared with binding in the unconstrained R state (−33.4 kcal mol −1 ) is called the cooperative free energy of binding (ΔG c = +6.3 kcal mol −1 ). To provide a molecular explanation for how O 2 binding is coupled to changes in tertiary and quaternary structure, Perutz developed his stereochemical model for cooperative O 2 binding, which is described in the following sections.
The High- and Low-Oxygen Affinity Quaternary Structures of Hemoglobin
The stereochemical mechanism proposed by Perutz identifies the T and R states within the MWC model as two discrete quaternary structures that differ in the relative orientation of the α1β1 and α2β2 dimers within the context of the Hb tetramer. The T-to-R quaternary switch of the tetramer involves about a 15-degree rotation of the α1β1 dimer relative to α2β2 (see Fig. 19-4, C ). The rotation occurs around an axis perpendicular to the C2 symmetry axis and passes through both α subunits. The β chains experience greater relative movement, moving closer together in the R state (as originally observed by Perutz). At the same time, the central cavity between the four globin chains, particularly the cleft between the β subunits, narrows in the R state and inhibits binding of 2,3-DPG, an important modulator of O 2 affinity (see 2,3-Diphosphoglycerate ).
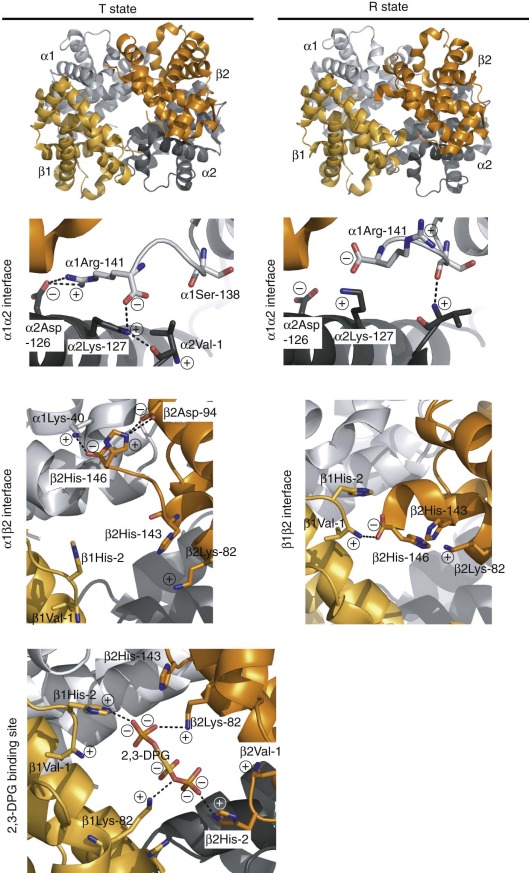
Interactions between α1β1 and α2β2 dimers (i.e., α1β2 interactions) are weaker in the R (oxy) state compared with the T (deoxy) state, which accounts for the observation that R-state tetramers dissociate into α1β1 dimers at low protein concentrations, whereas T state Hb remains a tetramer and emphasizes the role of α1β2 interactions in cooperative O 2 binding. In contrast there is little or no change in the α1β1- (or α2β2-) dimer interface between the T and R quaternary states. Consequently residues at these interfaces do not contribute significantly to allostery. Accordingly, free α and β monomers and α1β1 dimers bind O 2 noncooperatively with a high affinity similar to that of R-state tetramers. Conversely, when crystallized or embedded in a silica gel, T-state Hb cannot undergo a quaternary T→R transition and binds O 2 noncooperatively at low affinity. These latter findings demonstrate that interactions at the α1β2 interface reduce O 2 affinity in the T state and to a first approximation account for the tetramer’s cooperativity.
In tetrameric Hb, the α 1 chain contacts both the α 2 and β 2 chains. Each of these interfaces plays a role in the allosteric mechanism. Interactions between nonidentical chains (α1β2 and α2β1 contacts) account for the majority of the buried surface area. A more restricted set of α1α2 contacts are present in the T and R states. The β1β2 interface is present in the R state but is lost as the β chains move apart in the T structure.
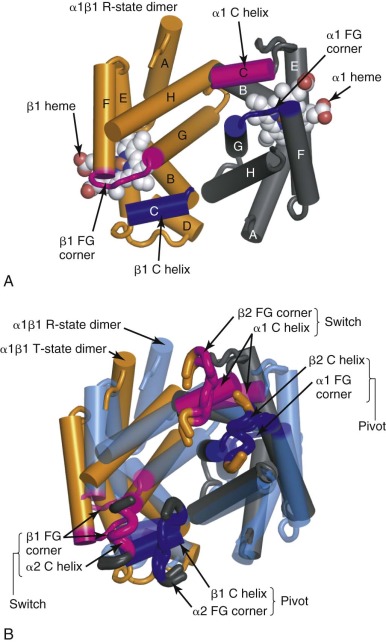
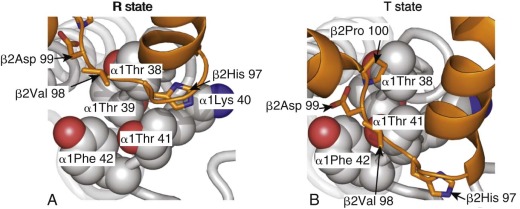
Shown in Figure 19-8, A , is the α1β1 dimer with the contact face for the α2β2 dimer facing up out of the page (the α2β2 dimer has been removed to reveal the contact surface). The α1β2 and α2β1 interfaces include the C helix and the FG corner (residues between the F and the G α helices) in each chain. These elements are colored blue and magenta in the α 1 and β 1 subunits shown in Figure 19-8 . The C helix from each Hb chain contacts the FG corner from the nonidentical chain across the α1β2 (or α2β1) interface. This interaction creates four pairs of contacts, as shown in Figure 19-8, B . In this panel, the C and FG segments of the α 2 and β 2 chains are shown as “bent tubes” overlaying the α1β1 dimer, with the rest of the chain omitted to allow the interface to be seen. Notably, the behavior of the C helix in α and β chains is different throughout the T→R quaternary change and is indicated by their different coloring in Figure 19-8 . Specifically, the molecular contacts between the C helices of the β chains (βC) and the FG corners of the α chains (αFG) change very little during the T→R quaternary transition (see Fig. 19-8, B , dark blue ). Therefore the βC–αFG contacts are known as the pivot (or joint ) contacts. In contrast, the αC helix moves relative to βFG corner, experiencing a translation of approximately 6 Å in the direction of the helix axis, equivalent to approximately one helical turn (5.4 Å) relative to the βFG corner (see Fig. 19-8, B , magenta ). The two alternative positions of the βFG loop relative to the αC helix are dictated by different side-chain packing interactions. Hence βAsp-99 in the βFG corner fits between the side chains of αThr-41 and αTyr-42 from the αC helix in the T state ( Fig. 19-9, B ). In the R state, βFG is shifted such that the side chain of βHis-97 fits between αThr-38 and αThr-41 in the αC helix (see Fig. 19-9, A ). These two alternative positions of the αC–βFG contact have led to this contact being described as the “switch” region of the α1β2 interface. It is thought that steric hindrance makes intermediates between these two positions unstable, giving rise to the two-state TR quaternary switch. The α1β2 interface is also characterized by different inter–subunit-hydrogen bonding patterns in the T and R states ( Fig. 19-10 ). The intricate nature of the switch region allows mutations at these residues to significantly perturb the stability of the T state ( L value in the MWC model) and therefore Hb function, sometimes with clinical consequences (see the second part of this chapter).
Although the switch region helps define distinct T and R structures, a range of R quaternary structures have been observed. For example, the C2 symmetry axis in liganded Hb that is crystallized under low-salt conditions at pH 5.8 is distinct from the T or R state structures determined at a neutral or high pH. This additional quaternary structure, R2 HbO 2 has been proposed to be either an intermediate in the T→R transition or a true end-point for fully oxy-Hb. The current understanding is that in the thermodynamic R state (fully oxy-Hb) proposed by the MWC model, Hb exists in a range of conformations, rather than as a single rigid structure. Crystal structures of the R, R2, and other quaternary variations provide information about the range of structures that are energetically accessible to HbO 2 . In support of this interpretation, recent measurements of O 2 binding to crystallized R- and R2-state Hb reveal that these are thermodynamically equivalent states, and a nuclear magnetic resonance (NMR) study of Hb in solution suggests that the average structure of liganded Hb lies approximately halfway between the crystallographic R- and R2-states. Despite considerable variation in the C2 symmetry axis between different R-state crystal structures, a set of common interactions between residues at the α1β2, α1α2, and β1β2 interfaces can be used as structural markers that identify the thermodynamic R state.
Altered Oxygen Binding at the Heme Site in the T and R States
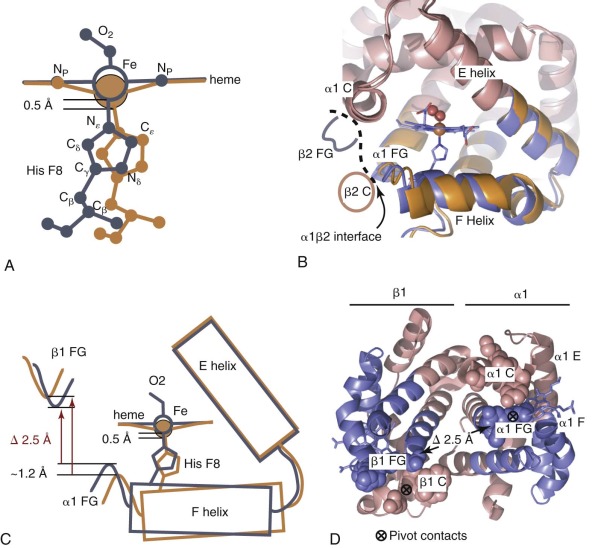
In the unliganded state, pentacoordinate heme iron (Fe[II]) makes four bonds to pyrrole nitrogens and one axial bond with proximal His F8 (α87 or β92). A water molecule sits in the distal pocket of the α chain but does not coordinate the heme iron. The distal pocket of β chains appears empty, or there is no discrete position for a water molecule. The heme site in T-state deoxy-Hb has a high-spin Fe(II) electronic configuration, and the iron is displaced 0.4 to 0.6 Å out of the plane of the four pyrrole nitrogen atoms in the direction of the proximal His F8 ligand. Studies on model porphyrins indicate that an out-of-plane iron is the preferred structure for high-spin Fe(II) porphyrin and hence the iron site in T-state deoxy-Hb is unstrained. Importantly, O 2 binding requires a change in electronic configuration to a low-spin, hexacoordinate, in-plane iron. This configuration is seen in R-state HbO 2 where the heme iron is only 0.09 and 0.06 Å out of the porphyrin plane in α and β chains, respectively. Hence the iron site in R-state HbO 2 is also unstrained. However, accommodating the O 2 -liganded heme in the T-quaternary structure introduces strain into the Hb structure because movement of iron into the porphyrin plane requires upward movement of the F-helix away from the α1β2 interface and consequently reduces the free energy of O 2 binding. Thus in effect, the low–O 2 affinity of the T state can be explained, because the O 2 -liganded heme is a poor fit in the T-state structure. In the R-state oxy-Hb, the His F8 imidazole ring is positioned over the heme such that the Fe–Nε bond is normal to the plane of the porphyrin ( Fig. 19-11, A , blue ). This geometry is important to minimize steric repulsion between the imidazole and porphyrin rings when iron is in the porphyrin plane. However, in the T-state structure, the His F8 imidazole is rotated relative to heme plane (see Fig. 19-11, A , orange ). In this position there is a greater energetic cost for moving iron into the heme plane because of steric repulsion between Cε of His F8 and the pyrrole nitrogens of the porphyrin.
Determining how strain, introduced into T-state Hb by O 2 binding, is distributed across the tertiary and quaternary T-state structure is the subject of ongoing research. In Perutz’s stereochemical model, the strain energy is at least partly compensated (or “stored”) by salt bridges present only in the quaternary T state, as described in the following sections.
Quaternary Interactions Contribute to Cooperativity
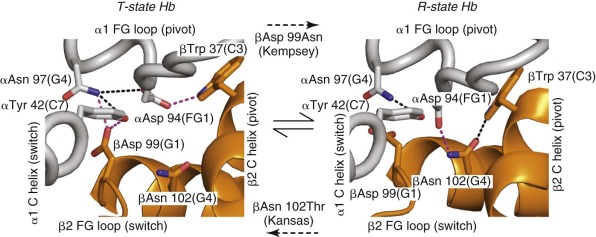
Salt bridges are electrostatic interactions between oppositely charged groups. These can involve the side chains of basic amino acid residues (Arg or Lys), acidic residues (Glu and Asp), N-terminal amino groups, or C-terminal carboxyl groups of polypeptide chains. Perutz identified eight specific salt bridges present in the T state (but not the R state) that stabilize the low-affinity quaternary conformation. More recent, higher-resolution crystal structures of T-state (deoxy-Hb) and R-state (oxy- and carbonmonoxy-Hb) Hb suggest minor modifications of this set of interactions ( Table 19-2 ). In the R state, these interactions are largely replaced by weaker hydrogen bonding interactions between polar residues or to water molecules. For example, the C-terminal residues αArg 141 (HC3) and βHis 146 (HC3) participate in T-state–specific salt bridges ( Fig. 19-12 ). Removing both αArg 141 and βHis 146 with carboxypeptidase B increases O 2 affinity and reduces cooperativity for the resultant modified Hb molecule. Similar conclusions were obtained using T-state Hb trapped in gels. Removal of αArg 141 alone caused a 15-fold increase in O 2 affinity of the T state, compared with a threefold increase for the β146 His→Leu mutation.
| Donor | Acceptor |
|---|---|
| T-State Quaternary Structure | |
| α1 Lys127 (H10) ε-amino group (+) | α2 Arg141 (HC3) carboxyl terminus (–) * |
| α1 Arg141 (HC3) guanidinium (+) | α2 Asp126 (H9) side chain carboxyl (–) * |
| α1 Lys40 (C5) ε-amino group (+) | β2 His146 (HC3) carboxyl terminus (–) * |
| β1 His146 (HC3) imidazolium (+) | β1 Asp94 (FG1) side chain carboxyl (–) * |
| α1 Arg92 (FG4) guanidinium (+) | β2 Glu43 (CD2) side chain carboxyl (–) * |
| α1 Asn97 (G4) side chain amine | β2 Asp99 (G1) side chain carboxyl (–) |
| α1 Tyr42 (C7) phenol | β2 Asp99 (G1) side chain carboxyl (–) |
| α1 Lys127 (H10) ε-amino group (+) | α1 Val1 (NA1) backbone carbonyl |
| β1 Val1 (NA1) amino terminus (+) | β1 Leu78 (EF2) backbone carbonyl |
| β1 Trp37 (C3) indole amino group | α2 Asp94 (FG1) side chain carboxyl (–) |
| R-State Quaternary Structure | |
| β1 Val1 (NA1) amino terminus (+) | β2 His146 (HC3) carboxyl terminus (–) * |
| α1 Asn97 (G4) side chain amide | α1 Tyr42 (C7) phenol |
| α1 Val1 (NA1) amino terminus (+) | α2 Ser138 (H21) backbone carbonyl |
| β1 Trp37 (C3) indole amino group | β1 Asn102 (G4) side chain carbonyl |
| β1 Asn102 (G4) side chain amine | α2 Asp94 (FG1) side chain carboxyl (–) |
| β1 Arg40 (C6) guanidinium (+) | α2 Thr41 (C6) hydroxyl |
| β1 Arg40 (C6) guanidinium (+) | α2 Leu91 (FG3) backbone carbonyl |
* Rows containing asterisks indicate salt bridges. No asterisk indicates hydrogen bonding interactions.
Beginning with Szabo and Karplus, several groups created mathematical models suggesting that T-state–specific salt bridges could account for the free energy of cooperativity. Consistent with experimental evidence, some models suggested that ligand binding in the T state strains but does not break the salt bridges. Importantly, it seems that the lower O 2 binding affinity of T-state Hb is the result of effects from salt bridges and other interactions at the α1β2 interface, which transmits ligand-induced strain in the allosteric core (heme, proximal His, F helix and FG corner). Crystallized T-state deoxyHb can be oxygenated without switching to the R structure. This procedure has revealed changes in the stereochemistry of the heme, shifts in the FG corners towards the R-state quaternary structure, and lengthening (i.e., weakening) of the T-state–specific salt bridges. For example, the C-terminal salt bridge between β 1 His 146 (HC3) and α 2 Lys 40 (C5) (see Fig. 19-12 ) is lengthened by 0.9 Å in the oxy T state compared to the deoxy T state. However, more recent computational approaches conclude that the free energy of cooperativity is also stored across many other molecular interactions.
Hydrogen bonds at the α1β2 interface may also be strained when the T state is liganded. For example, Hb variants with nonconservative switch-region substitutions at βAsp 99 or αTyr 42 show increased O 2 affinity and reduced cooperativity (see Fig. 19-10 and discussion of Hb Kempsey in High–Oxygen affinity Hemoglobin Variants). In contrast, conservative substitutions of Tyr or Asp gave less marked effects. Similarly, at the α1β2 hinge region, the side chain of βTrp37 makes an inter-subunit hydrogen bond in the T state that stabilizes the α1β2 interface in the T relative to the R state, and mutations of βTrp 37 decrease cooperativity.
Coupling of Oxygen Binding to Structural Changes in Hemoglobin
Superposition of the α1β1 dimers from T- and R-state Hb reveals a set of residues that have invariant tertiary structure. A group of 74 residues are used as a reference frame to describe structural changes in the α and β chains, as well as the α1β1 and α2β2 dimers, during T→R shifts. These include α-chain residues 23 through 42 (helices B and C), α 57 through 63 (helix E), α 101 through 111 (helix G), and α 118 through 125 (helix H); and β-chain residues β 51 through 57 (helix D), β 110 through 116 (helix G), and β 119 through 132 (helix H). These reference-frame residues predominantly map to the α1β1 interface, reinforcing the concepts that relative orientation of the α1β1 and α2β2 dimers represents the major quaternary rearrangement associated with cooperative O 2 binding.
The concerted changes in iron coordination and globin tertiary structure are shown diagrammatically in Figure 19-11 . During the T-to-R switch, helix F moves closer to the center of the globin, the gap between helices E and F narrows, and the heme iron moves toward the porphyrin ring (see Fig. 19-11, A -C ). In addition (not shown in Fig. 19-11 ) the α heme group moves about 0.5 Å towards the back of the heme pocket, and the β heme undergoes a larger translation of 1.5 Å and a 9-degree rotation. Motion of the heme iron in and out of the porphyrin plane upon O 2 binding and/or dissociation is transmitted through the proximal His, causing the F helix to pivot. The relatively rigid α-helical structure allows a small movement of the heme iron in or out of the porphyrin (≈0.5 Å) to be amplified, resulting in relatively large motions at the FG corners. The combined tertiary structural changes in the α and β subunits from the same high- affinity (α1β1 or α2β2) dimer bring the αFG and βFG corners approximately 2.5 Å closer together in the R state (see Fig. 19-11, D , blue ). In contrast, the relative positions of the αC and βC helices are little changed (see Fig. 19-11, D , pink ). The net result of contracting the distance between the FG corners, while keeping the distance between the C helices fixed, is that some of the C-to-FG contacts across the α1β2 interface must altered. As described previously, the α 1 C-to-β 2 FG contacts break and reform (switch), whereas the α 1 FG-to-β 2 C contacts pivot, resulting in the 15-degree rotation of α1β1 relative to α2β2 (see Fig. 19-4, C ). Although the αC helix (hinge region) does not undergo substantial conformation changes, it is believed to store significant strain upon T-state oxygenation.
Several lines of evidence confirm that the bond between iron and the proximal His-F8 plays key roles in transmitting the effect of O 2 binding to the F helix and in promoting structural rearrangements at the tertiary and quaternary level. For example, when deoxy-Hb was crystallized in the T-quaternary state and the crystals were oxidized and soaked with sodium cyanide (CN), the binding of the strong CN ligand to the T-state α chains forced the iron-His–F8 bond to rupture, indicating that introducing ligands into the T-state structure places the iron-His–F8 bond under strain. In another experiment, Ho, Barrick, and colleagues specifically ablated the iron-His–F8 bond using an F8 His-to-Gly mutation and introduced free imidazole, which bound into the proximal heme pocket and coordinated the heme iron. They found that this complex exhibited increased O 2 affinity and reduced cooperativity. This experiment suggested that the proximal heme ligand in wild-type Hb transmits approximately two-thirds of the cooperative free energy of O 2 binding. Steric interactions between the pyrrole rings and heme pocket residues are also involved.
Changes in Tertiary Structure Precede Quaternary Switching
Gibson, Eaton, and their colleagues employed time-resolved spectroscopy to study internal ligand movement and conformational events that occur after laser photolysis of the Fe–CO bond but before bimolecular rebinding of the ligand from solvent. This approach has shown that tertiary structural changes after ligand binding or dissociation occur within a much shorter time frame (hundreds of nanoseconds [ns] to 1-2 microseconds [µs]) compared with quaternary structural changes (on the order of milliseconds [ms]). Computational methods indicate that fast tertiary T→R structural transitions precede slow quaternary switching and that tertiary structural changes induced by oxygen binding to multiple subunits increases the probability that Hb will make the T→R switch. These studies show that the “concerted” quaternary switch, integral to the MWC model, is more accurately viewed as a coupling between rapid tertiary and slow quaternary events. Efforts to develop quantitative biophysical models to fully explain the coupling of tertiary and quaternary structural changes to ligand binding, incorporating all equilibrium O 2 binding and kinetic data, are ongoing.
Heterotropic Effectors of Oxygen Binding to Hemoglobin
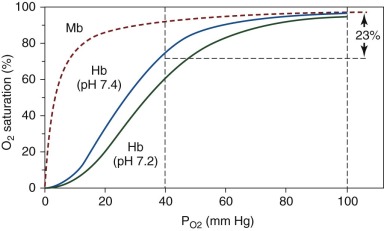
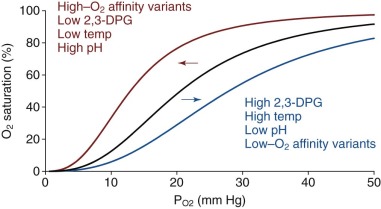
In addition to O 2 , other small molecules and ions known as heterotropic allosteric effectors bind with different affinities to T- and R-state Hb and as a result impact cooperative O 2 binding ( Box 19-3 ). The physiologically important species are CO 2 , hydrogen cation (H + ), Cl − and 2,3-DPG. In general, these molecules bind more tightly to the T state, thereby reducing the O 2 binding affinity of Hb. This binding causes a “right shift” in the O 2 saturation curve ( Fig. 19-13 ). Heterotropic effectors have their most significant effect at low P O2 values, because they augment O 2 release in the tissues. In the lungs, at relatively high P O2 (≈95 mm Hg), the slightly decreased O 2 affinity caused by allosteric effectors has little effect on O 2 uptake.
- •
Allosteric regulation refers to changes in the structure and activity of a protein that results from binding of regulatory substances outside of the protein’s active site.
- •
Allosteric regulators of Hb modulate O 2 binding by affecting the T-to-R transition.
- •
Cooperative O 2 binding is a form of allosteric regulation.
- •
Other allosteric regulators of Hb include H + , Cl − , and 2,3-DPG, which bind outside of the heme pocket to alter Hb O 2 affinity.
- •
CO 2 produced in peripheral tissues generates H + , which binds Hb to reduce its O 2 affinity, facilitating tissue oxygenation. This is the alkaline Bohr effect.
- •
2,3-DPG, a by-product of glycolysis, binds Hb to reduce its O 2 affinity. Hypoxia stimulates 2,3-DPG production to enhance O 2 delivery to tissues.
- •
Fetal Hb (α 2 γ 2 ) exhibits low affinity for 2,3-DPG, which facilitates transfer of O 2 from mother to fetus.
2,3-DPG, 2,3-Diphosphoglycerate; Cl − , chloride anion; CO 2 , carbon dioxide; H + , hydrogen cation; Hb, hemoglobin; O 2 , oxygen.
Carbon Dioxide Transport and the Bohr Effect
In 1904 the physiologist Christian Bohr showed that CO 2 reduced the O 2 affinity of Hb. Inside RBCs, carbonic anhydrase converts dissolved CO 2 to carbonic acid, which dissociates to bicarbonate and H + (i.e., CO 2 + H 2 O ⇌ H 2 CO 3 ⇌ HCO 3 − + H + ). Upon conversion from the R to the T state, ionizable chemical groups within Hb bind H + ion. Hence increasing [H + ] or decreasing pH shifts equilibrium in favor of the low-affinity T state. The decreased O 2 affinity as pH changes from about 7.4 in the lungs to 7.2 or less in tissue capillary beds is known as the alkaline (or physiological ) Bohr effect. Below pH 6.0, the affinity for O 2 increases again. This is called the acid Bohr effect, which does not occur in vivo. The physiologic importance of the Bohr effect is that increased pCO 2 (acidification) occurs at sites of high metabolic activity, promoting O 2 release from Hb where it is most required. For example, pCO 2 rises from approximately 35 torr in lung alveoli to about 50 torr in exercising muscles, causing the pH of blood to fall from about 7.4 to 7.2 or lower. This translates into a right-shifted O 2 saturation curve (see Fig. 19-13 ) with a 25% increase in the P 50 for O 2 by the alkaline Bohr effect (∂ logP 50 /∂ pH = −0.5 in 0.1 M Cl − , pH 7.4).
Hb facilitates the transport and disposal of CO 2 in several ways. As O 2 is released in actively respiring tissue capillaries, T-state Hb takes up H + , which promotes conversion of more CO 2 to bicarbonate via deprotonation of carbonic acid in red cells, thereby increasing the CO 2 carrying capacity of blood. Approximately 70% to 80% of CO 2 transport occurs as bicarbonate. In the lungs, the formation of R-state Hb releases bound H + , converting bicarbonate back to carbonic acid and CO 2 , which exchanges with air in the alveoli. An additional 15% to 20% of CO 2 is carried as carbamino-Hb, which is formed by reaction of CO 2 with the terminal amine groups of α and β chains (CO 2 + Hb-NH 2 ⇌ Hb-NH-COO − + H + ). Finally, about 7% of CO 2 gas is simply dissolved in the blood.
The Bohr effect is largely attributed to formation of specific salt bridges involving chemical groups that can bind and release H + in response to physiological changes in pH. His residues have basic side chains with pKa of about 6.5, meaning that they can adopt different charge states in the physiological pH range, depending on their chemical environment. In the equilibrium His + -to-His + H + reaction, equal amounts of the charged and neutral His side chains are present when pH equals pKa. When pH is greater than pKa, the neutral state predominates, and when pH is less than pKa, the positively charged state predominates. A key point is that local electrostatic interactions have a significant effect on the pKa value. Salt bridges in the T or low-affinity quaternary conformation stabilize the charged His + side chain, increasing its pKa. In other words, the affinity of these His side chains for H + is higher in the T state compared with the R state (the necessary condition for an allosteric effector). Then these salt bridges are broken during the transition to the R state when O 2 binds, causing a decrease on pKa and loss of the His protons.
The side chain that contributes most significantly to the alkaline Bohr effect is βHis-146 (HC3), which forms a salt bridge with βAsp-94 (FG1) in T-state Hb (see Fig. 19-12 ). Ho and colleagues measured proton dissociation by NMR to determine the pKa of all His side chains in T- and R-state Hb. They found that the pKa of βHis-146 increases from 6.4 in the R state to 7.9 in the T state and accounts for 60% of the Bohr effect (similar to the finding of Kilmartin et al ). Meanwhile, the sum of the contributions from all 13 surface histidyl residues in the αβ dimer accounted for 86% of the alkaline Bohr effect in 0.1 M Cl − , pH 7.4, the remaining effect being the result of other basic groups including the N-terminal amines. A tertiary Bohr effect has also been described (independent of quaternary bonds), which may involve protonation of buried His side chains including βHis-103 and the distal His side chains in the α and β heme pockets.
Cl − is another physiologic inhibitor of O 2 binding that plays a role in the Bohr effect. As bicarbonate ions move out of the RBC down a concentration gradient, they exchange for Cl − , which is imported into the red cell to maintain charge balance. This exchange is called the Hamburger or chloride shift, and is mediated by the anion exchange protein Band 3 on the erythrocyte plasma membrane. The alkaline Bohr effect is reduced by approximately 50% in the absence of Cl − . Virtually all of the chloride-independent contribution of the Bohr effect occurs through βHis-146. Multiple basic groups contribute to the chloride-dependent Bohr effect. The pKa of the amino terminal group of αVal-1 (NA1) and a number of His side chains show O 2 -linked pKa changes that are dependent on Cl − concentration. Specific Cl − binding sites may exist, but these have not been reliably identified through crystallographic studies. A more global network of electrostatic interactions of Cl − with multiple His side chains, the amino termini, and the positively charged central channel may ultimately be involved.
2,3-Diphosphoglycerate
2,3-DPG (also known as 2,3-bisphosphoglycerate or 2,3-BPG ) is produced from 1,3-DPG as a by-product of glycolysis in a reaction called the Rapoport-Luebering shunt. The concentration of 2,3-DPG in RBCs is about 5 mM, approximately equimolar with Hb tetramers. 2,3-DPG is an allosteric inhibitor of O 2 binding and increases the P 50 of HbA to achieve more efficient O 2 delivery. 2,3-DPG carries 5 negative charges that interact with cationic groups lining the entrance to the central Hb cavity between the amino termini of the β chains in the 2,3-DPG binding site (see Fig. 19-12 ). The 2.5 Å crystal structure of HbA containing bound 2,3-DPG reveals the major electrostatic interactions to be salt bridges between the βHis-2 and βLys-82 side chains and the carboxyl and phosphate groups of the effector (see Fig. 19-12 ). βHis-143 also lies in the binding pocket, but this side chain appears to neutral at pH 7 and may not be essential for 2,3-DPG binding. In the R-state structure, movement of the β chains closes the central channel and occludes that binding site for 2,3-DPG. Thus 2,3-DPG binding to Hb causes a large shift in equilibrium towards the T state (15- to 30-fold increase in the MWC parameter, L = [T 0 ]/[R 0 ], and a significant decrease in K T , see Allostery and the Perutz–Monod, Wyman, Changeux Model of Hemoglobin Function ). Kinetic and equilibrium measurements at O 2 binding suggest that up to 50% of the decrease in P 50 caused by 2,3-DPG is the result of changes in K T and perhaps even K R .
2,3-DPG provides a means for physiological regulation of Hb O 2 affinity under conditions of hypoxia that occur at high altitude. Similarly 2,3-DPG synthesis increases in anemia, favoring O 2 dissociation to counter decreased Hb concentration (see Fig. 19-13 ). In pregnancy 2,3-DPG can be increased by 30%. Fetal Hb (HbF, α 2 γ 2 ) shows weak binding to 2,3-DPG compared with HbA, which is presumably an adaptation to the P O2 gradient between the maternal and fetal tissues. A number of sequence changes in the N- and C-termini of γ globin, compared with β globin, have been investigated with the conclusion that the diminished 2,3-DPG binding is complex and cannot be attributed to single residue changes. HbF also shows limited CO 2 binding as carbamates but retains the chloride independent Bohr effect.
Glycolytic enzyme defects affect 2,3-DPG production and therefore HbO 2 dissociation. In general, glycolytic defects affecting enzymes downstream of 2,3-DPG such as pyruvate kinase result in a higher level of 2,3-DPG, causing a right-shifted O 2 dissociation curve with increased O 2 unloading by red cell HbA in peripheral tissues (see Fig. 19-13 ). Clinically this reduces the erythropoietic drive. In contrast, glycolytic defects upstream of 2,3-DPG such as hexokinase deficiency cause a reduction in 2,3-DPG concentration and lead to increased Hb O 2 affinity and increased erythropoietic drive. The physiologic differences exhibited in anemic glycolysis defective patients with right-shifted (pyruvate kinase deficiency) versus left-shifted (hexokinase deficiency) O 2 -saturation curves are described in a classic study from Oski et al. The patient with hexokinase deficiency (left-shifted curve) demonstrated rapidly decreased central venous P O2 tension and increased cardiac output in response to exercise. In contrast, the patient with pyruvate kinase deficiency (right-shifted curve) was able to offload more O 2 to tissues, reflected by a more gradually decreased central venous P O2 and a relatively slower rise in cardiac output. Thus at the same Hb levels a patient with pyruvate kinase deficiency (increased 2,3-DPG) has greater exercise tolerance than one with hexokinase deficiency, in which 2,3-DPG production is inhibited.
Redox Reactions of Hemoglobin
Physiologically, iron can exist in the II, III, and IV oxidation states. However, only Hb containing Fe(II) heme is capable of reversible binding to molecular O 2 . Oxidation of the heme-iron to the III and IV states can trigger a range of reactions that lead to Hb denaturation and the production of reactive oxygen species (ROS) that damage erythroid cells.
Autoxidation
In the presence of O 2 , α and β chains can spontaneously oxidize in a process called autoxidation, generating Fe(III) methemoglobin (met-Hb) and superoxide. The resulting Fe(III) met-Hb is unable to bind to O 2 , exhibits ~60-fold decrease in resistance to denaturation, is prone to form toxic insoluble globin precipitates containing dissociated hemin, and contributes to iron redox cycling and the generation of ROS (see Section on Production of ferryl heme and Hb protein radicals ). To counter this, erythrocytes contain high levels of superoxide dismutase and catalase that convert superoxide to H 2 O and O 2 . Erythrocytes also have high levels of the reducing agents ascorbate and glutathione, as well as a soluble form of cytochrome b5 reductase (also known as met-Hb reductase) that converts met-Hb back to the Fe(II) state. Despite these mechanisms, approximately 1% of Hb exists in the non-functional, oxidized Fe(III) state at any time, probably due in part to reactions with NO. The mechanisms of spontaneous Hb autoxidation have been the subject of much investigation. Early studies suggested that an electron might is transferred from Fe(II) to bound O 2 , leading to direct dissociation of superoxide anion :
Hb − Fe ( II ) − O 2 → Hb − Fe ( III ) + + O 2 · −
The normal Fe(II)–O 2 bond involves partial transfer of electron density from the iron to the O 2 , such that the HbO 2 can be described as ![]() . This charge transfer is reversed when molecular O 2 dissociates. The activation barrier for dissociation of negatively charged O 2 ·− from the positively charged iron center makes the barrier to direct superoxide anion dissociation high. An alternative reaction is an outer sphere electron transfer from the Fe(II)–porphyrin to an O 2 molecule that is not coordinated to the iron:
. This charge transfer is reversed when molecular O 2 dissociates. The activation barrier for dissociation of negatively charged O 2 ·− from the positively charged iron center makes the barrier to direct superoxide anion dissociation high. An alternative reaction is an outer sphere electron transfer from the Fe(II)–porphyrin to an O 2 molecule that is not coordinated to the iron:
Hb − Fe ( II ) + O 2 → Hb − Fe ( III ) + + O 2 · −
However, these reactions by themselves do not explain the dependence of autoxidation on O 2 concentration. It can be shown that both reactions cause autoxidation rate to increase up to a maximum asymptotic value with increasing [O 2 ]. However, a plot of autoxidation rate against O 2 concentration is bell-shaped, with the maximum rate occurring at approximately P 50 and an asymptotic value at very high O 2 concentrations. Work with various globins and model heme compounds suggested that electron transfer can occur by an outer sphere reaction for hexacoordinate Fe(II)-porphyrin that has a solvent molecule or other anionic ligand (L) at the sixth coordination site to facilitate formation of stable ferric iron complex :
Hb − Fe ( II ) − O 2 + L ⇌ Hb − Fe ( II ) + L + O 2 ⇌ Hb − Fe ( II ) − L + O 2 → Hb − Fe ( III ) + − L + O 2 · −
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


