- 16.1 Introduction
- 16.1.1 Mechanisms of immune destruction
- 16.2 Autoimmune haemolytic anaemias
- 16.2.1 Warm antibody haemolytic anaemias
- 16.2.2 Cold antibody haemolytic anaemias
- 16.2.3 Drug-induced autoimmune haemolytic anaemias
- 16.2.4 Paroxysmal nocturnal haemoglobinuria
- 16.2.5 Alloantibodies causing anaemia
- 16.2.1 Warm antibody haemolytic anaemias
- 16.3 Immune thrombocytopenia
- 16.3.1 Autoimmune immune thrombocytopenia
- 16.3.2 Drug-induced thrombocytopenia
- 16.3.3 Neonatal thrombocytopenia
- 16.3.1 Autoimmune immune thrombocytopenia
- 16.4 Immune neutropenia
- 16.5 Haematopoietic progenitor cells
- 16.6 Immune disorders of coagulation
- 16.6.1 Primary antiphospholipid antibody syndrome
- 16.6.2 Other antibodies to coagulation factors
- 16.6.1 Primary antiphospholipid antibody syndrome
- 16.7 Blood transfusion
- 16.7.1 Principles of blood transfusion
- 16.7.2 Risks of blood transfusion
- 16.7.1 Principles of blood transfusion
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
16.1 Introduction
In this chapter, those haematological diseases in which the immune response plays a pathogenic role are considered. For example anaemia, thrombocytopenia, neutropenia or disordered blood clotting can all be due to antibodies directed against components of blood. In most cases, these antibodies are autoantibodies. However, disease also results from the stimulation of alloantibodies (isoimmune antibodies) by repeated blood transfusions or pregnancy. Direct activation of complement by erythrocytes and the role of the immune system in bone marrow failure (such as aplastic anaemia) are also discussed here. Malignancies of lymphocytes, namely leukaemias and lymphomas, are discussed in Chapter 6.
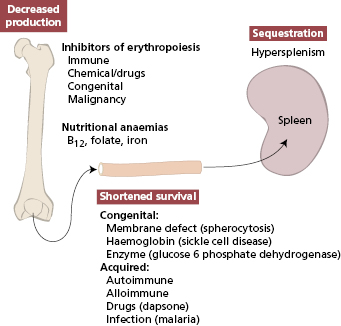
Reductions in circulating cells in general may be due to either failure of production or excess immune destruction (as illustrated for anaemia in Fig. 16.1).
16.1.1 Mechanisms of immune destruction
The immune system can destroy mature erythrocytes, platelets and neutrophils as well as some haematological precursors in the bone marrow. Immune destruction of red cells is used as the model shown later, though the mechanisms are common to all forms of destruction of cellular blood components resulting in cytopenias (Box 16.1).
- Antibodies attach to antigen on cell surfaces prior to phagocytosis in spleen – most common
- Complement-mediated lysis following antibody binding – less common
- Direct complement lysis without antibody involvement – rare
- Soluble immune complexes binding via CR1 (C3b) receptors (immune adherence) prior to phagocytosis
- Soluble immune complexes binding via Fc receptors (innocent bystander destruction) prior to phagocytosis
16.2 Autoimmune haemolytic anaemias
The common causes of anaemia are given in Fig. 16.1, which shows that the immune system is not often involved; nutritional deficiencies account for many more cases of anaemia than autoimmune processes. Autoimmune haemolytic anaemia (AIHA) is the commonest cause of shortened survival of red cells in Caucasians, though hereditary defects (such as sickle cell anaemia) are more common in other racial groups.
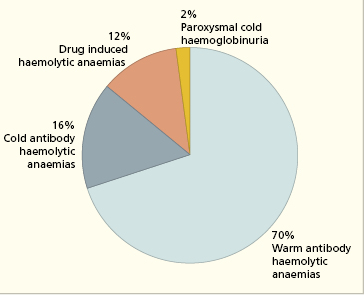
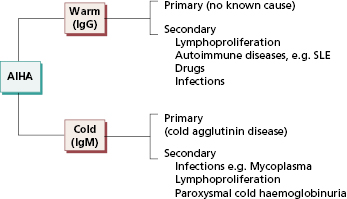
AIHA may be primary (idiopathic, with no known cause) or secondary to pre-existing disease. Autoantibodies formed in the secondary cases do not appear to be any different, either serologically or immunochemically, from those in primary AIHA. Fig. 16.2 shows the different types of AIHA.
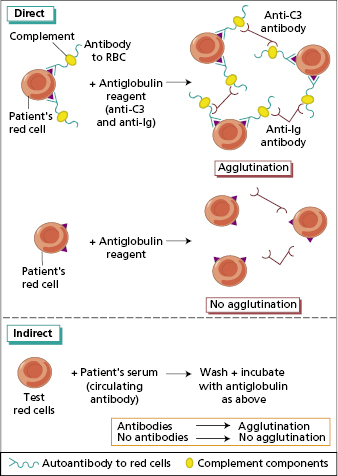
The diagnosis of AIHA depends on the demonstration of autoantibodies attached to the patient’s red cells or free in the serum. The screening test used is the Coombs’ test (Fig. 16.3); antibodies and complement components are detected on the surface of red cells by means of an antiglobulin reagent. This is a mixture of antibodies that reacts with IgG, IgM or C3 but cannot distinguish between specific antibodies directed against red cells and immune complexes attached to the surface by Fc or C3b receptors. In practice, the only immune complexes that are adsorbed sufficiently firmly to be a problem are drug–antibody complexes. Therefore, if the patient has signs of increasing haemolysis and no history of medication, a positive Coombs’ test is good presumptive evidence for AIHA due to autoantibodies. Specific antibodies to IgG, IgM and C3 can be used at different incubation temperatures to type the AIHA (Box 16.2) and antibodies eluted off the red cell surface to enable typing of their specificity.
- Warm reactive IgG autoantibodies, which are best detected at 37°C
- Cold reactive IgM autoantibodies, which are detected at temperatures between 4°C and 37°C
- Drug-provoked immune haemolytic anaemias
- Complement-activating IgG of paroxysmal cold haemoglobulinuria (Fig. 16.4)
16.2.1 Warm antibody haemolytic anaemias
The warm antibody type of AIHA (Table 16.1) affects all ages and both sexes, although most patients are over 30 years old. It is of varying severity and may be transient or persist for years. About one-half of the patients have idiopathic disease (Table 16.1), but in the remainder the anaemia is secondary to lymphoma or autoimmune disease, especially systemic lupus erythematosus (SLE) (see Fig. 16.4). The aetiology of primary idiopathic AIHA is unknown, although there are sporadic reports of familial occurrences of AIHA.
Table 16.1 Comparative features of warm and cold autoimmune haemolytic anaemia (AIHA)
| Warm AIHA | Cold AIHA | |
|---|---|---|
| Age (in years) | 30+ | 60+ |
| Cause of symptoms | Chronic haemolysis | Peripheral microvascular obstruction, e.g. Raynaud’s phenomenon |
| Mechanism of anaemia | Opsonization and phagocytosis | Intravascular haemolysis related to cold |
| Jaundice | Common | Uncommon |
| Splenomegaly | Common | Uncommon |
| Underlying disease | Present in approx. 50% | Uncommon but accompanying Raynaud’s phenomenon |
| Response to steroids | Good | Poor |
| Response to splenectomy | 50% of cases improve | Poor |
| Usual class of antibody + type of response | IgG – polyclonal | IgM – monoclonal/polyclonal |
| Commonest specificity of antibody | Usually non-specific | Anti-I antigen |
Usually, warm antibody AIHA is caused by IgG antibodies that react to protein antigens on the surface of red blood cells at body temperature.
Red cells from AIHA patients are direct Coombs’ test positive. The commonest reaction pattern (50%) on red cells is to have both IgG and C3 fixed on their surfaces; IgG only is found in 40% of cases. In the remaining 10%, complement alone is detected, nearly always in the form of C3d (see Chapter 1). The immunoglobulin is nearly always polyclonal, i.e. of mixed κ and λ light chain types.
Free autoantibodies can also be demonstrated in the serum of about one-third of patients by an indirect antiglobulin test (Fig. 16.3). A positive test for free autoantibody is associated with more severe haemolysis (as in Case 16.1). In most cases, IgG autoantibodies are non-agglutinating (and therefore called ‘incomplete’ by haematologists) but they are nevertheless destructive. If enzyme-treated cells are used, the sensitivity of the test is increased due to a reduction of surface charge, making the cells more ‘agglutinable’.
 Case 16.1 Primary autoimmune haemolytic anaemia
Case 16.1 Primary autoimmune haemolytic anaemiaA 32-year-old man gradually noticed that he had ‘yellow eyes’ and dark urine, felt continually tired and was short of breath when climbing stairs. He had no other symptoms; in particular, there was no itching, fever or bleeding; he was not taking any drugs. On examination, he was anaemic and jaundiced but afebrile, with no palpable lymphadenopathy, hepatosplenomegaly, rash or arthropathy.
On investigation, his haemoglobin was very low at 54 g/l. The white cell count appeared raised (40 × 109/l), but this was due to nucleated red cells being counted as leucocytes by the automated counter. The blood film showed gross polychromasia with nucleated red cells and spherocytes; the reticulocyte count in the blood was 9%. His serum bilirubin (47 mmol/l), aspartate transaminase (90 IU/l) and lactate dehydrogenase levels (5721 IU/l) were raised. Further tests showed that his red cells had IgG and C3 on their surfaces by the direct Coombs’ test. Serum contained warm non-specific autoantibodies (i.e. reactive with all the red cells in the test panel). Antinuclear antibodies and rheumatoid factor tests were negative and immunoglobulin levels were normal; there were no paraprotein bands in his serum or urine. Large amounts of urinary haemosiderin were detected.
A laboratory diagnosis of primary AIHA due to warm antibodies (leading to haemolysis and jaundice) was made. He failed to respond to high-dose corticosteroids and had a splenectomy 3 weeks later. Although impalpable, the spleen was twice the normal size; histology did not reveal a malignancy. He made a good post-operative recovery; his haemoglobin rose rapidly and the reticulocyte count fell. He took prophylactic penicillin for at least 2 years after surgery to prevent severe Strep. pneumonia infection.
Many antibodies can be eluted from the red cells’ surfaces even if there is no free antibody in the serum. These polyclonal autoantibodies may have specificity against a particular red cell antigen or represent a mixture of antibodies against common erythrocyte surface antigens.
The commonest pathogenesis in warm antibody haemolytic anaemia is coating of red cells by opsonizing antibody alone or with complement components (including C3b). Such opsonized cells are removed from the circulation by splenic macrophages (Box 16.1).
Management consists of attempts to reduce antibody production as well as excessive red cell destruction. Corticosteroids are the mainstay of treatment and have reduced mortality considerably. Other immunosuppressive drugs, such as cyclophosphamide and azathioprine, have been used as steroid-sparing agents. Unfortunately, the condition tends to relapse when azathioprine is stopped. There is now good evidence that the anti-CD20 monoclonal antibody Rituximab (Chapter 7) is useful in managing in AIHA resistant to conventional treatments. Removal of the B cells producing the autoantibodies by Rituximab is successful in approximately 70% of AIHA patients, but at the cost of destroying all mature B cells and resulting transient antibody deficiency for up to 1 year or so.
Splenectomy is nearly always beneficial if steroids fail; as well as removing the site of phagocytosis, a source of autoantibody production is eliminated. This has to be balanced against the increased risk of infection (see section 3.5.1). Blood transfusion is contraindicated unless anaemia is life threatening.
16.2.2 Cold antibody haemolytic anaemias
Cold antibody haemolytic anaemias may be primary or secondary (Fig. 16.4). Patients with cold haemagglutinin disease (CHAD) present with chronic haemolytic anaemia (anaemia, haemoglobinuria and jaundice) and severe Raynaud’s phenomenon on exposure to cold (see Table 16.1). Idiopathic CHAD is the most common form and is a disorder of elderly people; secondary cases occasionally occur in association with non-Hodgkin’s lymphoma, Mycoplasma pneumoniae infection or infectious mononucleosis. Rarely, a patient who has had ‘idiopathic’ CHAD for years develops a lymphoma.
Usually, cold antibody AIHA is caused by IgM antibodies that bind to polysaccharide antigens on the surface of red blood cells in the patient’s cold extremities. As the blood warms up again, complement is activated and intravascular haemolysis results. This is one of few known examples of a direct haemolytic role of complement in vivo. Red cells from all patients with CHAD have detectable IgM on their surfaces at 4°C; on warming, the antibody detaches from the erythrocyte surface, but fixed C3d can still be detected by a Coombs’ test. The temperature levels between which the antibody reacts with the red cell antigens is termed the thermal range.
Free cold autoantibodies (cold agglutinins) are also present in the patient’s serum. Ninety per cent of pathological cold antibodies are specific for I antigen (Case 16.2). This antigen occurs only on adult red cells and the ‘i’ antigen, by contrast, only on cord blood cells. Eight per cent of cold antibodies are anti-i; such cases are usually associated with infectious mononucleosis. In contrast to warm agglutinins (see Table 16.1), the cold antibodies found in idiopathic CHAD or in association with a lymphoma are monoclonal, though the amount of monoclonal antibody is usually far too small to be detectable as a paraprotein by serum electrophoresis; those that develop after an infection are polyclonal.
 Case 16.2 Cold haemagglutinin disease
Case 16.2 Cold haemagglutinin diseaseA 77-year-old man presented one winter with malaise and very cold hands and feet. He admitted to a tendency to bruise easily, and to passing dark urine in cold weather. He was not on any medication and was a non-smoker. On examination, he had some bruising on the shins and was mildly jaundiced. His fingers and toes were cold, but not ischaemic. He had small but palpable lymph nodes in both axillae and groins but no hepatosplenomegaly.
His haemoglobin was low (100 g/l) and the blood film showed rouleaux formation (autoagglutination) and polychromasia; neutrophil, lymphocyte and platelet counts were normal. He had raised serum bilirubin and lactate dehydrogenase levels: serum iron, folate and vitamin B12 measurements were normal. He had normal IgG (8.3 g/l) and IgA (1.2 g/l) levels and a slightly raised IgM (4.2 g/l); electrophoresis of serum and urine showed no paraprotein bands. He had a normal level of serum β2-microglobulin. There were cold antibodies in his serum that agglutinated red cells of ‘I’ specificity. A laboratory diagnosis of cold haemagglutinin disease leading to haemolysis and mild jaundice was made. He was advised to keep as warm as possible at all times. He has been seen regularly over the last 8 years but has not required active treatment or developed an overt lymphoid malignancy.
Treatment is usually unnecessary provided that the patient keeps the extremities warm. Steroid treatment and splenectomy are relatively ineffective since red cell destruction is predominantly intravascular. Treatment of an underlying lymphoma may stop the haemolysis, especially if Rituximab is used. Plasma exchange removes circulating IgM rapidly in severe cases.
16.2.3 Drug-induced autoimmune haemolytic anaemias
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


