Syndrome
Tumor
Risk (%)
Percent of malignancies that equal NHL
Ataxia-telangiectasia
Leukemia, NHL, HL, adenocarcinoma
>12
41–66
Common variable ID
NHL, gastric carcinoma
8–10
45–62
Severe combined ID
NHL, HL, leukemia, occupational adenocarcinoma
5
31–76
X-linked agammaglobulinemia
Leukemia, NHL, HL, adenocarcinoma
6
Wiskott–Aldrich
NHL, leukemia, HL
>10
59–75
Bloom
Leukemia, NHL, HL, adenocarcinoma
25
X-linked lymphoproliferative (after EBV infection)
NHL, HLH
24–35
Selective IgA deficiency
NHL, gastric carcinoma, thymoma, other
?
Etiology
Multiple genetic mutations within a single cell are required for malignant transformation. Like most neoplasms, lymphomas arise because of the inappropriate expression or loss of function of genes that regulate cell proliferation, differentiation, and programmed cell death (apoptosis). The malignant clone may result from increased proliferation of cells due to inappropriate expression of an oncogene such as c-myc, believed to be an important mechanism in high-grade pediatric lymphomas, or to alteration in genes, such as bcl-2, controlling apoptosis leading to decreased cell death, resulting in a more indolent clinical course, as is seen in adult low-grade lymphomas. The loss of a tumor suppressor gene expression and/or function, a mechanism common in many solid tumors, may also be seen in some patients with T-cell leukemia or lymphoma.
Viral infections may induce genetic changes and play a critical role in the pathogenesis of NHL. Examples of this include the role of EBV in the development of the genetic abnormalities associated with endemic (African) Burkitt lymphoma (BL) and that of human T-cell leukemia/lymphoma virus (HTLV) I and II in the development of adult T-cell lymphoma or leukemia. The oncogenic potential of EBV and HTLV is increased by infection early in childhood. The EBV genome is found in the DNA of tumor cells in 95 % of cases in endemic BL but in only 15 % of the sporadic cases [9, 10].
The distribution of endemic BL coincides with the geographical malaria belt in Africa. Malaria stimulates B-cell proliferation and T-cell suppression, and the interaction of this chronic effect with EBV, occurring at an earlier age than in developed countries, appears to be important in the pathogenesis of endemic BL [11]. The variation in incidence within this region, combined with case clusters and shifting foci of endemic BL, suggests the presence of additional cofactors such as concurrent infection with mosquito borne arboviruses and tumor promoters from local plant extracts [12]. The role of radiation in the development of lymphomas is not clear. Although there is an increased risk of NHL in patients treated with combined radiation and chemotherapy, there was no correlation of lymphoma incidence with increased radiation dose in atomic bomb survivors [13]. Another possible association of pesticide exposure and NHL remains unproven. A French study of household exposure to pesticides suggested that insecticide use during pregnancy and paternal household use of pesticide were associated with an increased risk of childhood acute leukemia and NHL [14].
Classification
Over the last 50 years, lymphoma classifications have evolved from being solely morphology-based. The Revised European–American Lymphoma (REAL) [15] and recent World Health Organization (WHO) classifications [16] define the subtypes of lymphoma by a combination of morphology, immunophenotype, cytogenetic abnormalities, and clinical presentation. The spectrum of pediatric NHL is considerably narrower than that of adult NHL, with the majority of childhood NHL being limited to four major categories (Table 48.2). By the WHO classification system, 50 % of children with NHL have Burkitt or Burkitt-like lymphomas, 20 % have T- or B-cell lymphoblastic lymphoma with T lineage predominating, 10 % have anaplastic large-cell lymphoma (ALCL), and 10 % diffuse large B-cell lymphoma (DLBCL), while the remainder are rare and unclassified subtypes [3]. Lymphoblastic lymphoma is morphologically and cytogenetically indistinguishable from acute lymphoblastic leukemia (ALL); these diseases are thought to represent a spectrum of a single disease, as reflected in the designation of T- or B-lymphoblastic leukemia/lymphoma in the WHO classification. An arbitrary cut-off of 25 % marrow involvement distinguishes stage IV NHL from ALL, a distinction of questionable biologic significance. Nonetheless, this differentiation remains important for the accurate comparison of the results of therapeutic trials. All other types of lymphoma, such as the many subtypes of nonanaplastic peripheral T-cell lymphomas comprise only 1–2 % of pediatric NHL [17] but form the majority of the less common T-cell subtype of PTLD.
Table 48.2
World Health Organization (WHO) classification of non-Hodgkin lymphoma commonly seen in childhood and adolescence
Type | Frequency | Subtypes |
|---|---|---|
Burkitt lymphoma | 40–45 % | •Burkitt lymphoma •Burkitt-like lymphoma (also called atypical Burkitt lymphoma) –B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma |
Lymphoblastic lymphoma | 20–25 % | •Precursor T-lymphoblastic lymphoma •Precursor B-lymphoblastic lymphoma |
Diffuse large B-cell lymphoma | 10 % | •Diffuse large B-cell lymphoma •T-cell/histiocyte-rich large B-cell lymphoma |
Anaplastic large-cell lymphoma | 10–15 % | •Mature/peripheral T-cell •Null-cell •Primary cutaneous anaplastic large-cell lymphoma |
Other | <5 % | •Primary mediastinal (thymic) large B-cell lymphoma with sclerosis •Peripheral T-cell lymphoma •Follicular lymphoma |
Pathology
The majority of pediatric NHL are diffuse, aggressive, high-grade lymphomas, which tend to arise in extranodal lymphoid tissue such as thymus or Peyer’s patches. The aggressive nature of most pediatric NHL is thought to reflect the growth rate of the counterpart normal lymphocyte precursor [18]. The stages of lymphocyte differentiation at which childhood lymphoma and leukemia are thought to arise [19, 20] are shown in Figs. 48.1 and 48.2.
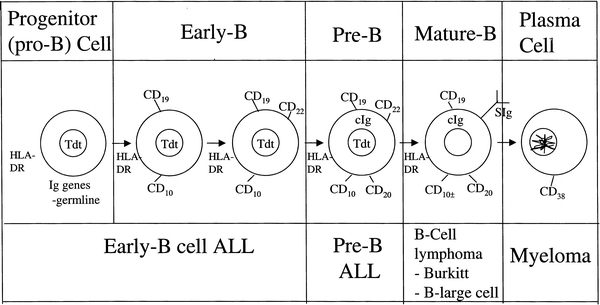
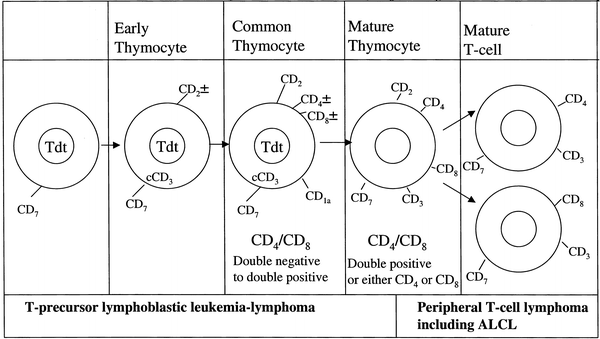
Fig. 48.1
Stages of B-lymphocyte differentiation
Fig. 48.2
Stages of T-lymphocyte differentiation. (Data from Rezuke WN, Abernathy EC, Tsongalis GJ. Molecular diagnosis of B- and T-cell lymphomas: fundamental principles and clinical applications. Clin Chem 1997;43:1814–23; and Goust JM, Jackson A. Lymphocyte ontogeny and membrane markers. Immunol Ser 1993;58:161–86.)
This rapid growth rate is reflected by the presence of numerous mitotic figures. Macrophages surrounded by a clear area, giving a starry sky appearance, may be seen in all four types of childhood lymphoma, although it is most commonly associated with BL. Follicular low-grade lymphomas rarely occur in children, and in patients less than 16 years of age tend to occur as localized disease, usually affecting the head and neck area. A population-based registry examined 25 cases of pediatric follicular lymphoma finding key differences from adult FL in that no cases had the classical translocation t(14;18) yet 55 % of cases expressed BCL-2, which did not have any prognostic significance in this series [21]. It has also been suggested that the absence of BCL-2 gives an improved prognosis. In addition to the four major types of NHL, children may present with a number of lymphoproliferative disorders, generally in the setting of congenital or acquired immunodeficiency.
Molecular Biology
The most common molecular alterations in NHL are rearrangements in immunoglobulin and T-cell receptor genes. Normally, rearrangement of immunoglobulin heavy and light chains and all four chains of the T-cell receptor (TCR) occurs at precise times during differentiation. In B cells, heavy chain rearrangement occurs before light chain and κ light chain before λ. In T cells, γ- and δ-chain genes rearrange before α and β. DNA polymerase chain reaction (PCR) methods are used to identify these immunoglobulin and T-cell receptor gene rearrangements and thereby establish clonality.
Rearrangements of these receptor genes are not always lineage-specific.
Immunoglobulin gene rearrangements may be seen in T-cell disease, and T-cell receptor rearrangements in B-cell disease. Rearrangements involving light chain receptor genes tend to be more specific for B-lineage disease than heavy chain gene rearrangements, which may also be observed in other lineages [22]. PCR measurement of specific Ig rearrangements is being utilized to detect minimal residual disease (MRD) after induction chemotherapy in B-NHL [23], while PCR measurement of the protein product of the t(2;5) translocation in blood and bone marrow is being evaluated as a measure of minimal disseminated disease (MDD) at diagnosis and MRD after initiation of therapy in patients with ALCL [24]. In T-LL MDD and MRD can be evaluated by PCR amplification of genetic abnormalities and clonal T-cell receptor rearrangements, but a recent COG study has suggested that the characteristic immunophenotype with coexpression of T-cell antigens and TdT, not found in normal blood and bone marrow, will allow MDD and MRD testing using flow cytometric methods [25].
Clinical Presentation
Pediatric NHL usually arises in extranodal lymphoid tissue, grows rapidly, and tends to spread early, so that the majority of children present with locally advanced or metastatic disease. Only 10 –15 % of childhood NHL present with primary peripheral nodal disease and a minority present with involvement of extralymphatic tissue such as bone, skin, lung, and gonad. Involvement of the CNS presents as leptomeningeal (headache, neck stiffness, back pain) or cranial nerve palsies. Paraplegia due to spinal cord compression also occurs. There is a clear link between the different subtypes of NHL and their clinical presentation.
Diagnosis
The aggressive nature of most pediatric NHL stresses the urgency to obtain a diagnosis and begin therapy. Correct diagnosis requires morphology, cytochemistry, immunophenotyping, cytogenetics, and molecular genetics. On occasion, fine-needle aspiration (FNA) from the tumor, or aspiration of pleural or pericardial fluid, may yield enough material for diagnosis and for limited additional studies such as cytogenetics or electron microscopy. In view of new biology-based approaches to therapy, however, fresh-frozen tissue is becoming increasingly important and excisional biopsies are often important to obtain adequate samples.
Before the patient is sent for biopsy, investigations to assess the extent of disease and of tumor-induced complications need to be made in a timely fashion. In particular, patients with large abdominal tumors need to be evaluated for gastrointestinal obstruction or perforation, as well as for renal impairment due to kidney infiltration, obstruction, or hyperuricemia. Patients with an anterior mediastinal mass need urgent evaluation for airway or superior vena cava (SVC) compression, right ventricular outflow compression, pleural effusion, and cardiac tamponade. It is important that these patients not be put into a supine position for the investigations, but instead procedures should be done with patients prone or lying on their side, and no patient with airway obstruction should be sedated for a procedure without consultation with an experienced anesthesiologist. When anesthesia is considered to be too hazardous and diagnosis cannot be obtained from a peripheral node, pleural/pericardial fluid or bone marrow, the mediastinal mass may be reduced in size by administration of corticosteroid therapy until a biopsy can safely be performed. An open biopsy must be performed as soon as possible, generally no longer than 24–36 h after starting steroids, so that sufficient tissue is available for pathologic diagnosis and for important biology studies. A single-institutional review of 23 children with mediastinal lymphoma who received prebiopsy steroids showed that 5 of 23 (22 %) had a delay in diagnosis, failure in definitive diagnosis or inaccurate staging as a result [26]. Appropriate prophylactic measures, such as the use of hydration and agents directed at reducing the risk of urate crystal formation, should be started early since steroid therapy may result in tumor lysis syndrome (TLS).
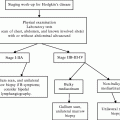
Staging
A comprehensive assessment includes blood cell count and differential, blood chemistry for renal and liver function studies, uric acid, potassium, phosphate, and calcium (tumor lysis syndrome) and lactate dehydrogenase (LDH) as a measure of tumor burden. Studies of the primary tumor include chest X-ray, computed tomography (CT) of neck, chest, abdomen and pelvis (and occasionally head), ultrasound and echocardiogram. Magnetic resonance imaging (MRI) scans are done for CNS and paraspinal disease. Staging work-up with functional imaging such as FDG-PET is increasingly being performed as studies demonstrate its increased sensitivity over conventional imaging methods [27], although limitations exist especially within the pediatric population. Extension of disease is determined by CSF examination and bilateral bone marrow aspirate and biopsy. Tests for CMV, VZV, hepatitis viruses, and EBV are recommended prior to therapy. HIV status should be assessed if clinically indicated.
The most widely used staging system for pediatric NHL is the St. Jude’s system (Table 48.3), which is applicable to all major subtypes and separates patients with localized disease (stages I and II) from those with advanced disease (stage III). Patients with stage IV NHL have bone marrow involvement with less than 25 % blasts and/or have CNS disease; more than 25 % tumor cells in the bone marrow is, by the WHO classification scheme, called leukemia. CNS disease is defined as leptomeningeal infiltration or isolated cranial nerve palsy. Isolated extradural disease which does not confer the same prognosis should be defined as stage III lymphoma, as should multifocal bone disease. Significant differences in intensity and length of therapy as well as in prognosis for patients with localized and advanced-stage NHL emphasize the importance of accurate disease staging.
Table 48.3
St. Jude’s staging system for pediatric non-Hodgkin lymphoma
Stage I | A single tumor (extranodal) or single anatomical area (nodal) with the exclusion of the mediastinum or abdomen |
Stage II | A single tumor (extranodal) with regional node involvement Two or more nodal areas on the same side of the diaphragm Two single (extranodal) tumors with or without regional node involvement on the same side of the diaphragm A primary gastrointestinal tract tumor, usually in the ileocecal area, with or without involvement of associated mesenteric nodes only, grossly completely resected |
Stage III | Two single tumors (extranodal) on opposite sides of the diaphragm Two or more nodal areas above or below the diaphragm All the primary intrathoracic tumors (mediastinal, pleural, thymic). All extensive primary intra-abdominal disease, unresectable All paraspinal or epidural tumors, regardless of other tumor sites Multifocal bone (now considered stage III rather than IV) |
Stage IV | Any of the above with initial central nervous system and/or bone marrow involvement |
For risk stratification of mature B-NHL, a more useful clinically-based grouping system was developed by the Lymphome Malins de Burkitt (LMB) group [28]. Group A (low-risk) defined as resected stage I and abdominal completely resected stage II; Group C (high-risk) defined as bone marrow disease (≥25 % L3 blasts) or CNS disease or both; Group B (intermediate-risk) comprises all others. In addition, an elevated LDH level at diagnosis that is above twice the upper limit of normal [28] or ≥ 500 U/L [29] continues to have negative prognostic significance among intermediate-risk patients and may be used to stratify LMB Group B patients into high- and low-risk strata in future studies.
Treatment Approaches and Prognosis
Surgery is occasionally needed to deal with acute complications such as intussusception, bleeding, or bowel perforation but most importantly to obtain adequate tissue for diagnosis. Localized abdominal tumors seen at the time of laparotomy are often easily resected, and the prognosis is excellent with a short course (6 weeks) of chemotherapy. Surgery should not be performed for the purpose of resection or for debulking of tumors; in addition, surgical interventions that delay the onset of chemotherapy should be avoided. The place of surgery in the assessment of residual tumors postchemotherapy is controversial. The French Society for Pediatric Oncology (SFOP) demonstrated that two-thirds of residual abdominal masses were necrotic [30] and suggested that second-look surgery was necessary to define remission status. The success of autologous transplant in patients with a slow response to primary therapy suggests that an accurate assessment of residual tumor may be useful and prognostic.
Radiation therapy (RT) has been relegated to a secondary role in the treatment of NHL. In patients treated with adequate chemotherapy, RT has been shown to increase both short-term and long-term morbidity without providing therapeutic advantage. RT is therefore omitted in most modern protocols except for patients with CNS disease. Even for patients presenting with a paraspinal mass and paraplegia, no advantage of addition of RT has been demonstrated, and RT should be limited to patients who fail to respond rapidly to chemotherapy [6]. Possibly because of rapid cell cycling, radiation given in single daily fractions is relatively ineffective in Burkitt lymphoma [31], although a later study suggests that hyperfractionation with at least three fractions per day may have some benefit [32]. This should be taken into account when total body irradiation (TBI) is considered for relapsed BL.
Combination chemotherapy is the primary treatment modality for childhood NHL. The treatment protocol should be based on the subtype and extent of disease. Prior to the start of therapy, measures must be taken to prevent TLS [33]. In BL, tumor lysis may be present prior to the start of therapy and is more likely to occur after therapy than in the T-cell neoplasms because of the higher cell turnover in BL. High-volume fluids to establish good urine flow and either allopurinol to prevent urate production or urate oxidase to cause urate breakdown should be started immediately. In the presence of significant pleural or pericardial effusions, right ventricular outflow, or superior/inferior vena cava obstruction, hyperhydration may need to be given in the setting of an intensive care unit. It is important to attempt correction of preexisting abnormalities before the initiation of chemotherapy, and hemodialysis may be necessary even before therapy is started in some cases. Establishment of adequate urine flow is essential to prevent potentially fatal hyperkalemia; potassium should be avoided in intravenous solutions until the period of risk for tumor lysis is past [33].
Hyperhydration is continued until the risk of TLS has diminished, which is usually about 5 days. Hypocalcemia occurs as a consequence of hyperphosphatemia and should only be treated if symptomatic, in which case, intravenous calcium can be given with caution, given the risk of calcium–phosphate precipitation in the kidney. Allopurinol inhibits xanthine oxidase and the further production of urate but will not reduce the amount of urate already present and can lead to accumulation of the precursors xanthine and hypoxanthine, both of which can also precipitate in renal tubules. The advantage of urate oxidase is that it converts uric acid into allantoin, a highly soluble compound and, thus, dramatically decreases uric acid levels, without the need for urine alkalinization. Recombinant urate oxidase (rasburicase) has been shown to be highly effective in multicenter randomized trials in adult and pediatric patients to reduce the rate of TLS and renal failure [34]. The potential for acute hemolysis should be carefully monitored when using urate oxidase in patients with glucose-6-phosphate dehydrogenase deficiency.
Chemotherapy
The use of multiagent, intensive chemotherapy regimens has markedly improved survival for children with NHL. The selection of a chemotherapeutic regimen is based on the lymphoma histology and/or whether a B- or T-cell immunophenotype is identified. Intensity and duration of therapy are significantly different for localized and advanced disease.
Burkitt and Burkitt-Like Lymphomas
Pathology
Burkitt lymphoma (BL) represents the neoplastic transformation of a relatively mature B-cell precursor that expresses surface immunoglobulin [35]. BL exhibits a diffuse monomorphic proliferation of cells with a high nuclear/cytoplasmic ratio and deeply basophilic cytoplasm containing lipid vacuoles. Single-cell necrosis with ingestion of debris by macrophages, leading to the starry sky pattern, is frequently present. The nuclei are similar to or slightly smaller than histiocyte nuclei, and the lymphoma cells have been categorized as small noncleaved lymphocytes. The cells in BL are fairly monomorphic in size and shape, with clumped or irregularly distributed nuclear chromatin and two to five nucleoli. In the Burkitt-like subtype, there is more heterogeneity in cell size, and nucleoli tend to be fewer and more prominent. There has been much controversy concerning the significance of Burkitt vs. Burkitt-like histology. Recent work from the BFM group, using gene expression analysis, is beginning to shed light on this distinction affirming that the majority of Burkitt-like lymphomas are molecularly identical to Burkitt lymphoma and can be treated with the same regimen (section in this chapter on Molecular Biology). Flow cytometry and immunohistochemistry demonstrate B-cell markers CD19 and CD20 and IgM class surface immunoglobulin with light chain κ or λ restriction. CD10 (common ALL antigen) may be present in some BL. Terminal deoxynucleotidyl transferase (TdT) is negative in Burkitt lymphoma cells, reflecting their mature phenotype.
Molecular Biology
BLs have characteristic cytogenetic changes, which in 80 % of cases involve translocation of the C-MYC proto-oncogene on chromosome 8 (involved in cell proliferation and differentiation) to the immunoglobulin heavy chain gene locus on chromosome 14 (t[8;14] [q24;q32]). In the remaining 20 % of cases, C-MYC is translocated to the κ light chain gene locus on chromosome 2 (t[2:8] [p11;q24]) or to the λ light chain locus on chromosome 22 (t[8;22] [q24;q11]). Although the t(8;14)(q24;q32) is common to both sporadic and endemic BL, the breakpoints within the two genes are different. Both chromosomal translocations result in juxtaposition of C-MYC next to the highly active Ig gene, leading to deregulation and uncontrolled expression of the C-MYC protein [35]. Gene expression profiling can be used to classify mature B-NHL into BL and DLBCL with increased precision. With this technology, Burkitt-like lymphoma was found to group with histologically classical BL based on similar gene expression signatures and together comprising 63 % of pediatric B-NHL [36]. Cytogenetic abnormalities have not been used for risk stratification, but emerging data suggest that rearranged MYC (8q24), del 13q, +7q) [37, 38] and 13q and 22q [39] are predictors of poor prognosis among B-NHL.
Clinical Presentation
Ninety percent of BL present with abdominal disease, with the remainder arising from B lymphocytes in the Waldeyer ring lymph nodes (tonsils, adenoids). Despite a similar histologic appearance, the sporadic and endemic BL differ in their clinical presentation. The peak age for presentation is 7 years in the African (endemic) type and 11 years in the sporadic type, with a male predominance (2:1–3:1) in both [40].
In North America and Europe, BL arises most commonly from relatively mature B cells in Peyer’s patches within the gastrointestinal (GI) tract, most commonly at the ileocecal junction. Gonadal and kidney involvement is common. Bowel obstruction is a common presenting symptom. Eighty percent of patients present with an abdominal mass or with abdominal pain, abdominal distension, nausea, and vomiting. A localized BL may act as the lead point for an intussusception, and the tumor is not infrequently diagnosed during surgery for acute appendicitis. Involvement of tonsil or adenoid may lead to airway obstruction, commonly associated with nontender cervical adenopathy.
Jaw involvement occurs in only 15 % of patients with sporadic BL and then usually as part of multiple bone metastases, whereas endemic (African) BL commonly presents with jaw tumors (70 %) as well as involvement of the GI tract and kidneys. Peripheral lymph node involvement is unusual in patients with the endemic form of the disease. Bone marrow and CNS involvement may be seen with BL. Endemic BL patients more commonly present with CNS disease (leptomeningeal, cranial nerve palsies, and paraplegia), whereas bone marrow disease occurs more frequently in patients with sporadic BL.
BL is an extremely fast growing malignancy with a cell cycle doubling time of 2–3 days and a high spontaneous rate of apoptosis [40]. Patients with these tumors have the highest risk of any malignancy of developing TLS before or after the start of therapy. The survival of patients with BL was very poor until chemotherapy protocols were designed to take advantage of the very rapid cell cycling.
Treatment
Treatment of Localized Burkitt Lymphoma
Survival of patients with localized BL and B-large-cell lymphomas exceeds 85 % at 5 years of follow-up, permitting investigators to focus on reducing treatment-related morbidity [6, 41].
In the Pediatric Oncology Group (POG) 9219 trial, patients with stage I and II nonlymphoblastic disease treated with 9 weeks of cyclophosphamide–doxorubicin-vincristine–prednisone (CHOP)-based chemotherapy achieved an event-free survival (EFS) close to 90 % and demonstrated that radiation therapy can be safely omitted even for bone disease [41]. A similarly excellent outcome was achieved by the international FAB/LMB 96 study with a 4-year EFS of 98 % and overall survival (OS) of 99 %, after two courses of COPAD chemotherapy (Fig. 48.3) [42]. It is difficult to compare efficacy of the two protocols as the POG trial enrolled patients with stage I and II resected and unresected disease, whereas the FAB/LMB96 study enrolled only patients with resected disease (stage I or stage II); unresected disease was assigned to Group B. It appears that either strategy is acceptable for localized B-NHL. Regardless of the exact therapy, the patients who relapsed did so within a year from diagnosis. Central nervous system prophylaxis is no longer recommended for patients with localized abdominal lymphoma, including those with BL histology.

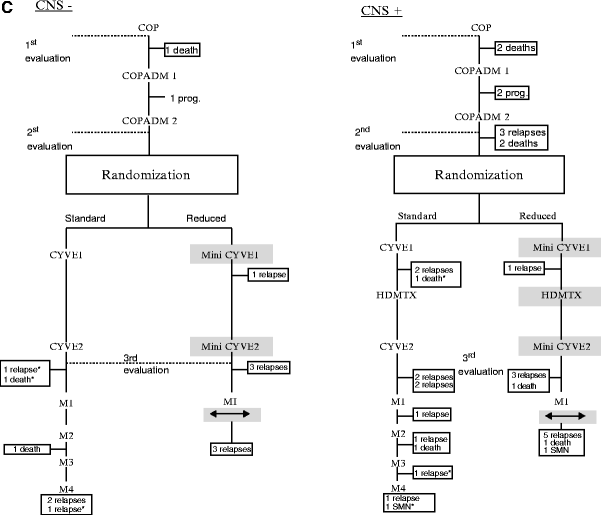
Fig. 48.3
FAB LMB 96 protocol for the treatment of childhood non-Hodgkin B-cell lymphomas, by risk group. (a) Group A therapy. (b) Group B therapy. (c) Group C therapy. (Reproduced with permission from: (a) Gerrard M, Cairo MS, Weston C, et al. Excellent survival following two courses of COPAD chemotherapy in children and adolescents with resected localized B-cell non-Hodgkin’s lymphoma: results of the FAB/LMB 96 international study. Br J Haematol 2008;141:840–7; (b) Patte C, Auperin A, Gerrard M, et al. Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-Hodgkin lymphoma in children and adolescents: it is possible to reduce treatment for the early responding patients. Blood 2007;109:2773–80; (c) Cairo MS, Gerrard M, Sposto R, et al. Results of a randomized international study of high-risk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood. 2007;109:2736–43.
Treatment of Advanced (Stage III and IV) Burkitt Lymphoma
During the last few decades major improvements have been seen in the cure rate of BL and mature B-ALL, from less than 30 % in the early 1980s to close to 90 % currently. Starting with the St. Jude Total B therapy [18], chemotherapy protocols have been developed based on the very rapid growth rate of these tumors; these include short-duration, dose-intensified, rapidly cycling courses involving high-dose S-phase drugs that cross the blood–brain barrier. Most successful protocols combine high doses of cyclophosphamide given in divided doses over a period of 3–5 days in combination with high-dose methotrexate (MTX) as well as high-dose cytosine arabinoside (Ara-C), together with vincristine and with or without anthracycline. All protocols include a corticosteroid in the form of prednisone or dexamethasone.
Reports of the best outcomes include those of the Société Française d’Odontologie Pédiatrique (SFOP) LMB 89 protocol (Table 48.4). A total of 561 patients were allocated to three risk groups (A, B, and C) and therapy was adapted to each risk group. Therapy for Group C patients (more than 25 % blasts in the bone marrow and/or CNS disease) consisted of 7 months of intensive therapy, which included high-dose MTX (8 g/m2 per dose) and high-dose Ara-C (3 g/m2 per dose). Cranial irradiation (24 Gy) was given to patients with CNS disease. This protocol resulted in an OS of 91 % at 5 years, 87 % for stage IV, 88 % for B-ALL, and an improvement in DFS for CNS-positive patients to 79 % from 19 % in the earlier LMB 81 study [28]. The 5-year DFS was 95 % for patients with a low LDH compared to 87 % for those whose LDH was increased twofold or more (p < 0.001), except in Group C patients in whom neither LDH nor bone marrow involvement was predictive of outcome. To build upon these excellent results, the SFOP, the Children’s Cancer Group (CCG), and the United Kingdom Children’s Cancer Study Group (UKCCSG) combined to conduct the French–American–British FAB/LMB 96 (Fig. 48.3) trial, which aimed at reducing therapy and minimizing toxicity while maintaining efficacy of LMB89. Among children with intermediate risk B-cell NHL (Group B), a reduction to half-dose cyclophosphamide (1.5 g/m2) in Induction Block 2 and the omission of Maintenance Block M1 yielded a 4-year EFS of 91 %, no different from the full dose arm [43]. Delay in therapy of >21 days between courses 1 and 2, however, significantly adversely impacted survival [44].
Table 48.4
Outcome for patients with advanced-stage Burkitt lymphoma
Protocol | Number | Stage or risk group | Outcome (%) | References |
|---|---|---|---|---|
LMB 89 | 420 | III/IV/B-ALL CNS+ | 87–93 % 5-year EFS 79 % 5-year EFS | Patte et al. [28]. |
FAB LMB 96 Group B | 451 | I/II unresected III/IV, CNS- | 98 % 4-year EFS 85–90 % 4-year EFS | Patte et al. [43]. |
FAB LMB 96 Group C | 204 | IV, BM+, CNS- IV, BM-, CNS+ IV, BM+, CNS+ | 88 % 4-year EFS 82 % 4-year EFS 61 % 4-year EFS | Cairo et al. [45]. |
BFM-95 | 283 | R1/R2 R3/R4 CNS+ | 94 % 3-year EFS 81–85 % 3-year EFS 70 % 3-year EFS | Woessmann et al. [46]. |
COG ANHL01P1 | 48 Group B 42 Group C | Group B, III/IV Group C, BM+/CNS+ | 93 % 2-year EFS 86 % 2-year EFS | Cairo et al. [48]. |
For Group C patients, the probability of 4-year EFS and OS was 79 % and 82 %, respectively, including patients on the reduced intensity arm. In patients who responded following COPADM2 and were randomized, the 4-year EFS was 90 % on the standard arm compared to 80 % on the reduced intensity arm [45]. Importantly, CNS-positive patients showed similar outcomes to LMB89 (EFS 75 %) after high-dose methotrexate (8 g/m2) and additional, intrathecal chemotherapy without cranial irradiation [45]. An important finding of this study was the delineation of groups with poorer outcomes who require intensification or alternative therapy in future protocols, such as those who responded poorly to the COP prephase (incomplete responders 78 % vs. nonresponders 30 % 4-year EFS), patients with combined bone marrow and CNS disease (61 % 4-year EFS) [45], and those with primary mediastinal B-cell lymphoma with sclerosis (72 % 4-year EFS) [43].
Other groups have produced similar results. In the BFM-NHL 86/90 and 95 studies, the BFM confirmed the safe omission of cranial radiation even for CNS-positive disease and the importance of high-dose methotrexate and high-dose Ara-C in advanced disease. They also confirmed that toxicity could be reduced and efficacy maintained by shortening intravenous methotrexate to 4 h vs. 24 h in those with limited stage B-NHL (Groups R1 and R2: stage I, II, and III with LDH < 500 U/L—failure-free survival (FFS) 95 %) but not in patients with advanced disease (Fig. 48.4 and Table 48.5) [46].
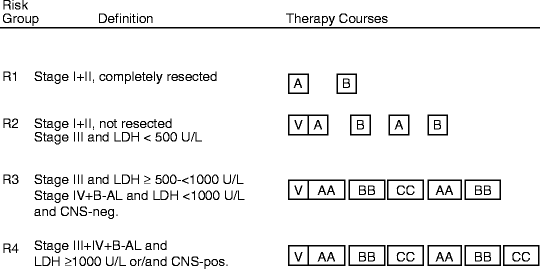
Fig. 48.4
BFM-95 protocol for treatment of B-cell NHL and anaplastic large-cell lymphoma. (Reproduced with permission from Woessmann W, Seidemann K, Mann G, et al. The impact of the methotrexate administration schedule and dose in the treatment of children and adolescents with B-cell neoplasms: a report of the BFM Group Study NHL-BFM95. Blood 2005; 105:948–58.)
Table 48.5
BFM-95 protocol for treatment of B-NHL
Drug | Dose | Day | ||||
|---|---|---|---|---|---|---|
1 | 2 | 3 | 4 | 5 | ||
Prephase V | ||||||
Dexamethasone orally/IV | mg/m2 | 5 | 5 | 10 | 10 | 10 |
Cyclophosphamide IV 1 h | 200 mg/m2 | x | x | |||
Methotrexatea IT | 12 mg | x | ||||
Cytarabinea IT | 30 mg | x | ||||
Prednisolonea IT | 10 mg | x | ||||
Course A | ||||||
Dexamethasone orally/IV | 10 mg/m2e | x | x | x | x | x |
Vincristine V | 1.5 mg/m2e | x | ||||
Ifosfamide IV 1 h | 800 mg/m2 | x | x | x | x | x |
Cytarabine IV 1 h | 150 mg/m2 | x–xg | x–xg | |||
Etoposide IV 1 h | 100 mg/m2 | x | x | |||
Methotrexate IVb | 1 g/m2 | x | ||||
Methotrexatea IT | 12 mg | x | ||||
Cytarabinea IT | 30 mg | x | ||||
Prednisolonea IT | 10 mg | x | ||||
Course B | ||||||
Dexamethasone orally/IV | 10 mg/m2e | x | x | x | x | x |
Vincristine IV | 1.5 mg/m2f | x | ||||
Cyclophosphamide IV 1 h | 200 mg/m2e | x | x | x | x | x |
Doxorubicin IV 1 h | 25 mg/m2 | x | x | |||
Methotrexate IVb | 1 g/m2 | x | ||||
Methotrexatea IT | 12 mg | x | ||||
Cytarabinea IT | 30 mg | x | ||||
Prednisolonea IT | 10 mg | x | ||||
Courses AAc, d | ||||||
Methotrexate IVb | 5 g/m2 | x | ||||
Methotrexatea IT | 6 mg | x | x | |||
Cytarabinea IT | 15 mg | x | x | |||
Prednisolonea IT | 5 mg | x | x | |||
Course CCc | ||||||
Dexamethasone orally/IV | 20 mg/m2e | x | x | x | x | x |
Vindesine IV | 3 mg/m2h | x | ||||
Cytarabine IV 3 h | 3 g/m2 | x–xg | x–xg | |||
Etoposide IV 2 h | 100 mg/m2 | x–xg | x–xg | xg | ||
Methotrematea IT | 12 mg | x | ||||
Cytarabinea IT | 30 mg | x | ||||
Prednisolonea IT | 10 mg | x | ||||
Both the BFM and LMB groups showed that patients with residual disease following three courses of chemotherapy could be successfully treated with high-dose chemotherapy (HDCT) and autologous bone marrow transplant (ABMT). In BFM-90, only 1 of 6 patients suffered from disease progression and in LMB-89, 9 patients out of 12 achieved a CR after HDCT and ABMT [28, 29]. By comparison, four of five partial responders in BFM-86 treated with chemotherapy alone died. These results suggest that surgery to confirm residual tumor is necessary, as high-dose therapy may be successful for tumors that respond slowly but remain chemotherapy-sensitive.
Although it is generally accepted that cranial radiation does not improve results, the outcome for CNS-positive patients remains inferior at 82 % compared to 88 % 4-year EFS for bone marrow-positive/CNS-negative patients, and 90 % 4-year EFS for the collective group randomized to standard treatment (Table 48.4) [45]. Further intensification of methotrexate is currently being tested for this subgroup.
The addition of the anti-CD20 monoclonal antibody rituximab to chemotherapy is standard for all adult B-NHL. Its value in pediatric CD20-positive NHL has not been definitively tested. In a recent BFM phase II window study in patients with newly diagnosed B-NHL and B-ALL, the overall response rate to one course of rituximab was 41 % with tolerable toxicity, suggesting that the antibody has efficacy even in very high-grade pediatric B-NHL [47]. A recently closed COG study demonstrated that rituximab can be added to Group B and Group C LMB-type therapy without increased toxicity (Table 48.4) [48]. Whether addition of rituximab to the successful intensive pediatric protocols will add to survival, however, remains to be proven.
Summary of the Findings of the Cooperative Group Trials
1.
Advanced-stage Burkitt and B-cell ALL can successfully be treated with a short duration (3–6 months) of very intensive, multiagent alkylator-based chemotherapy.
2.
Successive cycles of chemotherapy need to be given with minimal delay to prevent progression of these rapidly growing tumors.
3.
Improvement in cure rate in advanced-stage disease occurred with increasing intensity of chemotherapy, particularly higher doses of methotrexate.
4.
The addition of high-dose antimetabolite therapy, such as high-dose MTX and high-dose Ara-C, to intrathecal therapy is successful in prophylaxis and treatment of CNS disease, although CNS-positive patients still have an inferior prognosis compared to non-CNS-positive patients.
5.
Involvement of the bone marrow alone at diagnosis is no longer indicative of a poor prognosis. However, involvement of both CNS and bone marrow still carries an inferior prognosis.
6.
A high LDH level at diagnosis remains an adverse prognostic factor in patients on intermediate risk therapy but not in the most intensively treated patients.
7.
Most patients with BL who relapse will do so within the first 8 months after diagnosis. Patients with BL who survive disease-free for 10 months are likely to be cured.
8.
Patients with residual, histologically proven disease after three therapy courses have an increased risk of disease recurrence, which is not reduced by local surgery or radiation, but may be improved by intensification of chemotherapy or megadose therapy followed by hematopoietic stem-cell rescue.
All of these successful protocols are highly toxic, require frequent hospital admissions for treatment of toxicities, and require experience from oncologists in order to learn to give them without undue mortality. Studies are under way to attempt to reduce toxicity without changing efficacy. These include tailoring of therapy to risk of relapse, different methods of growth factor support, judicious reduction in chemotherapy doses and/or duration, and consideration of the addition of nontoxic synergistic therapy, such as monoclonal antibody therapy.
Lymphoblastic Lymphoma
Pathology
Lymphoblastic lymphoma (LL) consists of cells that are morphologically indistinguishable from acute lymphoblastic leukemia (ALL). It presents as a diffuse proliferation of medium-sized cells with a high nuclear/cytoplasmic ratio and scant amounts of basophilic cytoplasm (usually less basophilic than Burkitt lymphoma cells). The nuclear chromatin is finely stippled with indistinct nucleoli, and some degree of nuclear convolution may be seen in up to 50 % of cases, but this feature is not required for diagnosis and its presence does not seem to affect prognosis [49]. Mitotic activity is prominent and the starry sky histological pattern is commonly seen. The cells stain strongly for acid phosphatase; TdT is usually positive. By convention, LL that involves more than 25 % lymphoblasts in the bone marrow is called lymphoblastic leukemia and is indistinguishable from T-lineage ALL (T-ALL).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


