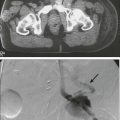
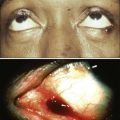
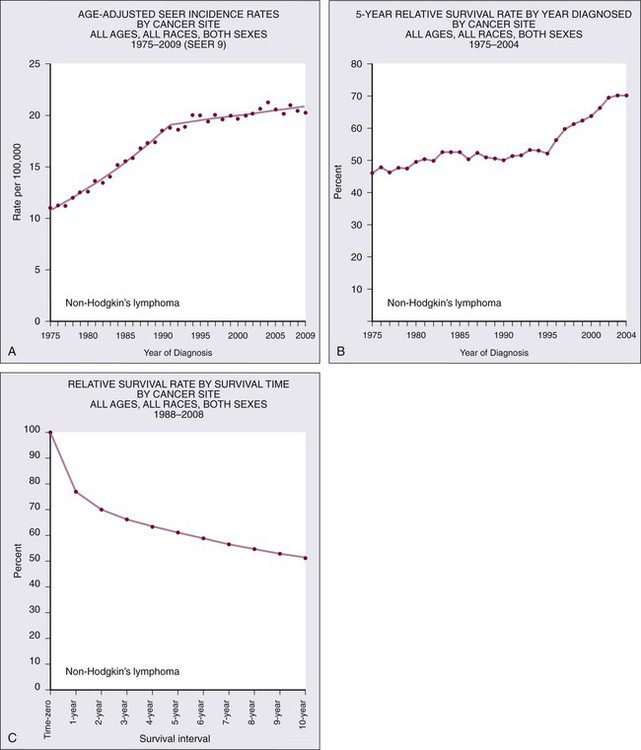
106 Mark J. Roschewski and Wyndham H. Wilson • Non-Hodgkin lymphoma (NHL) is the most common hematologic malignancy; more than 70,000 were estimated to be diagnosed in the United States in 2012. Diffuse large B-cell (DLBCL) and follicular lymphoma (FL) are the most common subtypes, each comprising approximately one third of cases in the Western Hemisphere. • In the Western hemisphere, approximately 85% of lymphomas are of B-cell origin and 15% are of T-cell/natural killer-cell origin. T-cell lymphomas have a higher incidence in parts of Asia. • Etiology of most lymphomas is complex and multifactorial. Defects in host immunity that increase the risk and infections associated with NHL include Helicobacter pylori, Epstein-Barr virus (EBV), human immunodeficiency virus (HIV), human T-cell leukemia/lymphoma virus type I (HTLV-I), hepatitis C, and human herpesvirus–8 (HHV-8). • Specific genetic abnormalities associated with lymphomas include translocations of BCL-2 (t(14;18)) in FL and DLBCL; BCL-6 (t(3;16)) in DLBCL; BCL-10 (t(11;18)) in mucosa-associated lymphoid tissue lymphoma; MYC (t(8;14), t(2;8), t(8;22) in Burkitt lymphoma and subsets of DLBCL; BCL-1 (t(11;14)) in mantle cell lymphoma (MCL); and ALK (t(2;5)) in anaplastic lymphoma kinase (ALK) + anaplastic large T-cell lymphoma (ALCL). • Gene expression profiling can subdivide the largest group of lymphomas, DLBCL, into three subtypes with distinct pathogenetic mechanisms and decidedly different prognoses. • Excisional biopsy is preferred for initial diagnosis and should be reviewed by an experienced hematopathologist. Fresh-frozen tissue should be collected for additional studies. • World Health Organization (WHO) Classification of Lymphoid Malignancies recognizes lymphomas that may be unclassifiable with morphologic features alone. Clinical and molecular features are essential for subclassification of lymphomas. • Staging evaluation includes patient history and physical examination; complete blood cell count and chemistry studies including lactate dehydrogenase; computed tomography (CT) of chest, retroperitoneum, and pelvis; and bone marrow biopsy. Positron emission tomography (PET) aids in the detection of extranodal disease and is more sensitive than CT alone. • Rituximab, a chimeric monoclonal antibody against CD20, is an essential component of the front-line and relapsed treatment of B-cell lymphomas. • Treatment of indolent lymphomas such as follicular lymphoma, marginal zone lymphoma, and lymphoplasmacytic lymphoma is for palliative benefit and is not curative; treatment is frequently curative for DLBCL, ALK+ ALCL, Burkitt lymphoma, and lymphoblastic lymphoma; and proper dose-intensity of therapy is essential. • MCL demonstrates remarkable clinical heterogeneity; no curative therapy exists, but a subset of patients can have a long indolent course without therapy. • Peripheral T-cell lymphomas (PTCLs) other than ALK+ ALCL have a poor prognosis and are difficult to cure with standard therapy. • High-dose therapy with stem cell transplant cures a smaller fraction of patients with relapsed aggressive lymphomas in the rituximab era. • Numerous novel agents that have been developed on the basis of biological targets of lymphoma subtypes are demonstrating significant promise. Non-Hodgkin lymphoma is the most common hematologic malignancy, with an estimated 70,130 cases diagnosed in the United States in 2012.1 One in 47 men and women will be diagnosed with NHL during their lifetime, with B-cell lymphomas representing 80% to 85% of those cases and T-cell lymphomas comprising the remainder. NHL is the seventh most common malignancy in the United States with a slight male predominance.2 Even though NHL affects all ages, its incidence increases steadily with each decade of life between ages 20 to 80, with a median age of 66. Whites have the highest incidence of NHL, followed by Hispanics, blacks, and Asians, and it is least commonly diagnosed in Native Americans/Alaska natives. From the early 1970s to the 1990s, the annual percent change in the incidence of NHL in the United States increased steadily at a compound rate of almost 4%, partly the result of infection with human immunodeficiency virus (HIV) and improved diagnostic techniques, but has been stable between 2004 and 2009 (Fig. 106-1A). NHL is the eighth most commonly diagnosed malignancy worldwide.3 It is more commonly diagnosed in developed areas, with the highest incidence rates in North America, Australia, and Europe and the lowest rates in Asia and the Caribbean.3 Outcomes have improved, with death rates from NHL declining recently. The 5-year relative survival rate for all cases of NHL increased to 70% in 2001 through 2007 compared with 51% in the 1980s and 47% in the 1970s; 10-year survival rates are now slightly above 50% (see Fig. 106-1B and C). Although the majority of people who die of NHL are older than 75 years, it remains the fourth most common cause of cancer-related death in persons aged 20 to 39 years.1 The sequence of events that results in NHL is unknown. Inherited genetic susceptibility and/or shared environmental exposures is suggested by registry-based studies that demonstrate slightly increased risk of developing NHL in first-degree relatives of patients with lymphoma and patients with monoclonal gammopathy of undetermined significance (MGUS).4,5 No germline mutations have been identified, and the majority of patients with NHL have no familial clustering. Many factors related to the host’s immune status combined with environmental exposures such as organic solvent exposure and wood products correlate with an increased risk of NHL (Box 106-1).6,7 Conflicting reports exist on the connection between exposure to ultraviolet (UV) radiation, vitamin D levels, and the lifetime risk of NHL.8 Rare, extranodal T-cell lymphomas are also encountered in the setting of immunologic defects. Enteropathy-associated T-cell lymphoma (EATCL) often occurs in the setting of celiac sprue, and hepatosplenic T-cell lymphoma (HSTCL) occurs in young men with a history of solid-organ transplantation or other immune defects.9 The current World Health Organization (WHO) classification of lymphomas recognizes a subset of diffuse large B-cell lymphomas (DLBCL) that arise as a consequence of chronic inflammation and are frequently associated with Epstein-Barr virus (EBV).10 The classic example is pyothorax-associated lymphoma, which was first reported in 1987 in patients treated for tuberculosis by artificial pneumothorax. Other cases of DLBCL may occur in the setting of chronic inflammation, such as chronic skin ulcers or osteomyelitis.11 The role of chronic immune stimulation is less ambiguous in infection-associated lymphomas, such as in lymphoma of mucosa-associated lymphoid tissue (MALT) in which the lymphoproliferation is frequently antigen driven.12–15 Evidence for the essential role of the chronic immune stimulation is that eradication of the underlying infection, such as chronic hepatitis C or Helicobacter pylori, can result in remission in many cases.18–18 Viruses may also function as co-factors in lymphomagenesis, whereby they may exert their effect through genomic integration, which leads to alterations in gene expression, and/or by directly affecting cellular proliferation. EBV, human T-cell leukemia/lymphoma virus type I (HTLV-I), and human herpesvirus–8 (HHV-8), in particular, have well-established oncogenic roles in subtypes of NHL.19–22 In the case of EBV, it appears to have a direct oncogenic role in lymphomas of immunocompromised patients, such as those infected with HIV and in the posttransplant period.11 HTLV-I has a direct role in adult T-cell leukemia/lymphoma (ATLL), but carriers have only a 2% to 5% lifetime risk of developing disease with a latency period of 30 to 40 years. HHV-8, also known as Kaposi sarcoma herpes virus (KSHV), has been associated with primary effusion lymphoma (PEL), which is a rare B-cell lymphoma that occurs primarily in highly immunosuppressed patients with AIDS who are often co-infected with EBV.23 The crucial step in ensuring an accurate pathological diagnosis is an adequate tissue biopsy. Attention should be paid to the biopsy site with the largest or most rapidly enlarging node, or functional imaging such as combined fluorodeoxyglucose-labeled positron emission tomography and computed tomography (FDG-PET/CT) may be used to guide biopsy sites.24 In most cases, fine-needle aspiration is insufficient for initial diagnosis because lymph node architecture is usually critical for diagnosis.25 Excisional lymph node biopsies are preferred, but multiple core needle biopsies can be adequate in situations in which involved nodes are not easily accessible. Clinicians must have a low threshold for re-biopsy in circumstances in which the original biopsy is nondiagnostic or ambiguous. Re-biopsy should be performed in most situations of suspected relapse because inflammatory conditions can radiographically mimic lymphoma and it is not uncommon for indolent lymphomas to transform their histology. Lymphomas are a heterogeneous group of diseases with a variety of natural histories. Histologic classification schemes have been developed to organize lymphomas into groups with shared pathogenesis and clinical behavior with an aim to guide treatment. The classification systems for lymphomas have changed frequently and dramatically since they were first introduced in the 1950s and continue to evolve with technologic advances and scientific discovery. These classifications have evolved from exclusively morphologic to the current working system that incorporates immunophenotype and hallmark genetic abnormalities. The most recent version of the WHO classification system in 2008 places great emphasis on molecular and cytogenetic abnormalities in conjunction with clinical variables to define individual subtypes (Box 106-2). In 1956, Henry Rappaport of the U.S. Armed Forces Institute of Pathology proposed a simple system of lymphomas based on the growth pattern of the disease (nodular vs. diffuse) as well as the predominant cell’s well-differentiated, poorly differentiated, undifferentiated, or histiocytic appearance. With the use of this system, nodular lymphomas composed of small lymphocytes were generally considered indolent disorders whereas so-called histiocytic lymphomas were aggressive. The Rappaport system was used until the 1970s when the American pathologists L.J. Lukes and R.D. Collins and the German pathologist Karl Lennert each proposed classification systems that reflected advances in cellular immunology, cell morphology, and lymphocyte lineage by dividing entities into B-cell and T-cell disorders on the basis of their cell surface markers.26,27 The immune phenotype proved fundamental for the accurate classification of lymphoma, with early studies showing that most lymphomas are of B-cell origin (Table 106-1), but these studies still did not address clinical concerns and were not uniformly embraced. Table 106-1 Typical Immunophenotype of Major Subtypes of B-Cell Non-Hodgkin Lymphomas In 1994, the International Lymphoma Study group developed a consensus list of diseases that could be recognized by pathologists and was codified in the Revised European–American Classification of Lymphoid Neoplasms (REAL) classification system; this ultimately became the WHO classification system and is the recognized standard today.28 The first WHO classification, published in 2001, defined diseases by four features: morphology, immunophenotype, molecular genetic characteristics, and clinical information,29 whereas the updated 2008 version places greater emphasis on distinctive immunologic and molecular profiles of lymphomas and emphasizes the importance of clinical characteristics.30 Insight into the genetic features that characterize tumor subtypes has aided in the diagnosis of lymphomas and identification of potential therapeutic targets. A variety of complementary technologies can measure the expression of thousands of genes on a solid platform, termed molecular profiling, and can link biology to a genetic expression signature. The application of gene expression profiling (GEP) to lymphoma has provided insights into unique molecular signatures of distinct types of B-cell malignancies31 and can relate lymphoid neoplasms to normal stages in B-cell development and physiology. This approach has provided a new way to classify lymphomas and predict clinical outcome. Molecular profiling has advanced the understanding of lymphomagenesis through identification of genes important in cellular proliferation, differentiation, and apoptosis.32–36 At a molecular level, genetic lesions identified in lymphomas include oncogene activation or loss of tumor suppressor genes caused by chromosomal translocation, deletion or mutation, or the integration of viral genomes (Table 106-2). Some lymphomas rely on the continuous signaling provided by these oncogenes, termed oncogene addiction, whereas others seem more reliant on signaling pathways not controlled by oncogenes, termed non-oncogene addiction.37 GEP has also helped define the molecular makeup of the nonmalignant cells in tumor specimens, the so-called microenvironment that is important for pathogenesis and may include potential therapeutic targets.38,39 Table 106-2 Major Molecular Translocations in Non-Hodgkin Lymphomas MALT, Mucosa-associated lymphoid tissue; GC, gastric cancer. Cases exist that cannot be classified with certainty. The term gray zone lymphoma was first introduced in 1998 for cases that shared morphologic and immunophenotypic features of Hodgkin lymphoma and DLBCL.40 It has been demonstrated that primary mediastinal B-cell lymphoma (PMBL) has an overlapping gene expression profile with nodular-sclerosing Hodgkin lymphoma (NSHL).41,42 Given that PMBL and NSHL exhibit clinical and biological overlap, the term mediastinal gray zone lymphoma (MGZL) was first used in 2005 by Traverse-Glehen and colleagues to identify those cases with features intermediate between PMBL and NSHL.43 The term has more than theoretical interest because MGZL has an inferior prognosis with contemporary therapies.44,45 As such, the 2008 version of the WHO classification system includes a provisional category of B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and classic Hodgkin lymphoma that includes these cases of MGZL. Similarly, the distinction between high-grade lymphomas such as DLBCL and Burkitt lymphoma (BL) can be challenging on morphologic and immunophenotypic grounds.46 The distinction between the two entities is critical, because patients with BL require high-intensity chemotherapy. Two studies have demonstrated that BL has a unique molecular profile and that a subset of cases classified as DLBCL have a BL molecular profile.47,48 It is now appreciated that between 8% and 10% of cases of newly diagnosed DLBCL will harbor a mutation in the MYC oncogene and have a poor outcome with R-CHOP (rituximab + cyclophosphamide, doxorubicin, vincristine, and prednisone).49,50 Some cases of MYC+ DLBCL may have dual translocations affecting both MYC and BCL-2, which portends an even worse outcome.51–54 The WHO 2008 classification includes a provisional category for these cases that is referred to as “B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma.” Accurate staging is essential for determining optimal management and prognosis. Initial evaluation begins with a history focused on the pace of signs or symptoms suggestive of lymphoma, the presence or absence of “B symptoms” (fevers, chills, drenching night sweats), possible sites of nodal and extranodal involvement, and assessment of possible underlying immunodeficiency. A history of exposure to pathogens including HIV, EBV, hepatitis C, and HTLV-I should specifically be addressed. Medication history is important, with emphasis on history of exposure to immunosuppressive agents such as chemotherapy, tumor necrosis factor-α inhibitors, or corticosteroids. When performing the physical examination, the physician should assess for hepatosplenomegaly and examine Waldeyer’s ring, epitrochlear nodes, and popliteal nodes, which may be difficult to measure on radiographic imaging. Extranodal involvement such as the skin should be closely examined. Laboratory tests should include a complete blood cell count (CBC) and serum chemistry with determination of lactate dehydrogenase (LDH) level. HIV and serologic tests for hepatitis B and C are needed regardless of reported exposure history. Viral tests such as for HTLV-I should be done in high-risk populations, whereas EBV viral loads may have prognostic value in specific lymphomas such as PTLDs and extranodal NK-cell/T-cell lymphoma, nasal type.55 Serum β2-microglobulin has prognostic implications for many lymphomas. Recommended tests that supplement the history and physical examination are listed in Table 106-3. Table 106-3 Evaluation of a New Patient with Non-Hodgkin Lymphoma The Ann Arbor staging system that was developed for Hodgkin lymphoma is the standard, but NHL does not spread along contiguous nodes as seen in Hodgkin lymphoma (Table 106-4). Contrast-enhanced CT of the chest, abdomen, and pelvis is standard for assessing sites involved with lymphoma. Lymph nodes are considered involved if the long axis is 1.5 cm or more or if the long axis is 1.1 cm or longer and the short axis is more than 1.0 cm. Lymph nodes that are less than or equal to 1.0 cm in both axes are considered uninvolved.56 Pitfalls in determining disease stage based on anatomic imaging alone is that enlarged nodes may not be involved with lymphoma, and extranodal sites of disease such as bone or skin involvement may be missed. Involvement of the bone is best evaluated by magnetic resonance imaging (MRI) and FDG-PET scans. A head MR image and lumbar puncture with evaluation of the cerebrospinal fluid (CSF) by cytology and flow cytometry57 should be performed in high-risk patients or if there are signs or symptoms suggestive of central nervous system (CNS) involvement. Patients at high risk for CNS involvement include those with extranodal involvement and those with highly aggressive lymphomas such as BL and precursor lymphoid neoplasms.49,50,58,59 Bone marrow involvement impacts both management and prognosis60 and should be assessed in all patients with NHL. It is not uncommon to find discordant lymphoma subtypes in the bone marrow and biopsy samples.61 Table 106-4 Non-Hodgkin Lymphoma: Ann Arbor Staging Classification and the Cotswold Modifications FDG-PET exploits the enhanced rate of glucose utilization in tumor cells compared with normal surrounding cells, a process known as the “Warburg effect.”62 FDG-PET provides a semi-quantitative measurement of tumor involvement in NHLs that has superior sensitivity to anatomic imaging.63 FDG uptake is dependent on several variables and is subject to interpretation. Nonetheless, FDG-PET is commonly used for staging of the disease of patients with newly diagnosed aggressive lymphomas, but its impact on treatment decisions is unclear. Clinical variables are powerful and independent predictors of outcome. A number of prognostic indices aid in the prognosis of individual NHL subtypes.64–67 An international project identified that inferior outcome was related to age older than 60 years, stage III or IV disease, serum LDH value above normal range, Eastern Cooperative Oncology Group (ECOG) performance status of 2 or higher, and involvement of two or more extranodal sites. A clinical prognostic model, termed the International Prognostic Index (IPI), was developed with these five factors and stratifies patients into quartiles with differing disease-free survival rate at 5 years (Table 106-5). The IPI is the standard for assessing prognosis in DLBCL, as well as for comparison between clinical trials. Because the IPI was developed before use of rituximab, a revised IPI was suggested, but it was not validated in a larger prospective study.68,69 Table 106-5 *One point is given for the presence of each of the following characteristics: age > 60 years, elevated serum LDH level, ECOG performance status ≥ 2, Ann Arbor stage III or IV, and more than two extranodal sites. †One point is given for the presence of each of the following characteristics: age > 60 years, elevated serum LDH level, hemoglobin level < 12 g/dL, Ann Arbor stage III or IV, and number of nodal sites ≥ 5. ‡Points are based on age, ECOG performance status, white blood cell count, and serum LDH level. Gene expression profiling (GEP) is also an important prognostic tool that demonstrates molecular heterogeneity within tumors. In DLBCL, for example, morphologically indistinguishable tumors show marked heterogeneity in gene expression and these patterns of expression correspond to the cellular origin of the lymphoma according to its stage of differentiation.70 DLBCL can thereby be divided into at least three different subtypes: germinal center B-cell (GCB)-like, activated B-cell (ABC)-like, and PMBL that arise by distinct pathogenetic mechanisms and have a very different prognosis.38,42,71,72 A molecular prognostic model of survival based on signatures of germinal center B cells, proliferating cells, reactive stromal and immune cells in the lymph node, and major histocompatibility complex class II cells, was developed for CHOP-treated DLBCL.71 Overall survival (OS) was superior in patients with the GCB-type compared with the ABC-type DLBCL, a finding that was independent of the IPI (Fig. 106-2). The distinction between GCB-like and ABC-like subtypes of DLBCL carries prognostic information in patients treated with rituximab-containing regimens.34 Wilson and associates demonstrated that treatment with dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab (DA-EPOCH-R) resulted in a 5-year progression-free survival rate (PFS) of 100% for GCB-like DLBCL compared with 67% for non–GCB-type DLBCL.73 In relapsed DLBCL, different responses were observed in bortezomib-containing regimens.74 The gene expression profiles of follicular lymphoma (FL) and mantle cell lymphoma (MCL) have also identified subgroups with different prognosis on the basis of cellular proliferation75 and tumor-infiltrating cells.39 GEP is impractical to perform on all patients with newly diagnosed lymphomas, so multiple immunohistochemical (IHC) models using combinations of antibodies have been developed to predict GCB and non-GCB subtypes of DLBCL.76–80 The concordance rate with GEP is variable, however, ranging between 74% and 93%.81 The “Tally” algorithm in which results are not analyzed in a specific order demonstrated the best concordance with GEP at 93% (Fig. 106-3). IHC models will require further validation and standardization but have the advantage of being widely available. MicroRNAs (miRNA) are short noncoding regions of the RNA that regulate gene expression and have been used to discriminate between normal cells and cancer cells.82 Recently a 9-miRNA signature was shown to separate cell lines derived from GCB and ABC-like subtypes of DLBCL.83 MiRNA profiling demonstrates promise in predicting outcomes in DLBCL,84 FL,85 and MCL86 and will require further study and validation. Accurate assessment of response to therapy is critical, particularly in curable lymphomas. Treatment response should be documented by physical findings, and all abnormal tests should be repeated. End-of-therapy assessment is usually performed 3 to 6 weeks after completion of therapy unless progression is suspected earlier. An International Working Group (IWG) workshop held under the auspices of the National Cancer Institute in 1998 standardized response criteria based on the bi-dimensional measurements of involved nodal groups.87 The spleen is considered to be a nodal site and is assessed by CT. Focal lesions in the liver are considered measurable. Patients with bone marrow involvement before initiating therapy are required to be morphologically free of lymphoma to be considered in complete remission. The response criteria were updated in 2007 and state that if all involved nodes have regressed to 1 cm or less, symptoms of disease have disappeared, and bone marrow biopsy is without involvement, the patient is in complete remission.56 Because residual necrotic tissue and inflammatory cells may prevent bulky nodes from reducing to 1 cm or less, FDG-PET can help distinguish residual tumor cells from tissue necrosis. The timing of the imaging after therapy affects the positive predictive value, and it is recommended that FDG-PET be performed 3 to 8 weeks after chemotherapy and 8 to 12 weeks after radiation therapy.88 In patients with bulky mediastinal lymph nodes, FDG-PET confirmation of a complete response is essential because these nodes frequently do not completely regress.
Non-Hodgkin Lymphoma
Epidemiology and Risk Factors
Incidence, Distribution, and Death Rates
Risk Factors and Predisposing Conditions
Diagnosis and Classification
Classification of Lymphomas
History of Lymphoma Classification Systems
Follicular
MALT
Marginal Zone, Nodal
Mantle Cell
Diffuse Large B-Cell
Mediastinal Large B-Cell
Burkitt
CHARACTERISTIC IMMUNOPHENOTYPE
CD20+, CD3−
CD10+, CD5−
CD20+, CD3−
CD10−, CD5−
CD23−
CD20+, CD3−
CD10−, CD5−
CD23−
CD20+, CD−
CD10−, CD5+
CD24−, PRAD1
CD20+, CD3−
CD20+, CD3−
CD20+, CD3−
CD10−, CD5−
TdT−

Molecular Genetics of Non-Hodgkin Lymphoma
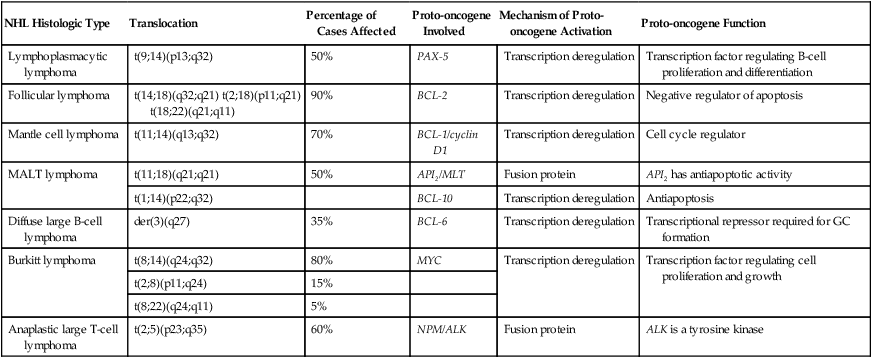
NHL Histologic Type
Translocation
Percentage of Cases Affected
Proto-oncogene Involved
Mechanism of Proto-oncogene Activation
Proto-oncogene Function
Lymphoplasmacytic lymphoma
t(9;14)(p13;q32)
50%
PAX-5
Transcription deregulation
Transcription factor regulating B-cell proliferation and differentiation
Follicular lymphoma
t(14;18)(q32;q21) t(2;18)(p11;q21) t(18;22)(q21;q11)
90%
BCL-2
Transcription deregulation
Negative regulator of apoptosis
Mantle cell lymphoma
t(11;14)(q13;q32)
70%
BCL-1/cyclin D1
Transcription deregulation
Cell cycle regulator
MALT lymphoma
t(11;18)(q21;q21)
50%
API2/MLT
Fusion protein
API2 has antiapoptotic activity
t(1;14)(p22;q32)
BCL-10
Transcription deregulation
Antiapoptosis
Diffuse large B-cell lymphoma
der(3)(q27)
35%
BCL-6
Transcription deregulation
Transcriptional repressor required for GC formation
Burkitt lymphoma
t(8;14)(q24;q32)
80%
MYC
Transcription deregulation
Transcription factor regulating cell proliferation and growth
t(2;8)(p11;q24)
15%
t(8;22)(q24;q11)
5%
Anaplastic large T-cell lymphoma
t(2;5)(p23;q35)
60%
NPM/ALK
Fusion protein
ALK is a tyrosine kinase
Gray Zone Lymphomas
Staging and Prognosis
Principles of Evaluation and Staging
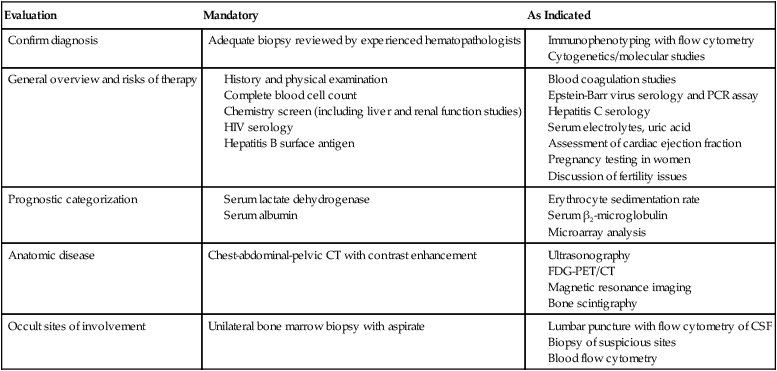
Evaluation
Mandatory
As Indicated
Confirm diagnosis
Adequate biopsy reviewed by experienced hematopathologists
General overview and risks of therapy
Prognostic categorization
Anatomic disease
Chest-abdominal-pelvic CT with contrast enhancement
Occult sites of involvement
Unilateral bone marrow biopsy with aspirate
Stage
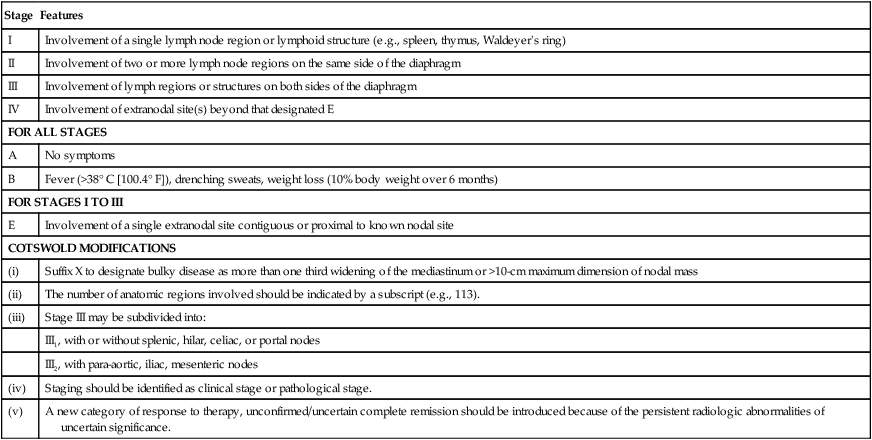
Features
I
Involvement of a single lymph node region or lymphoid structure (e.g., spleen, thymus, Waldeyer’s ring)
II
Involvement of two or more lymph node regions on the same side of the diaphragm
III
Involvement of lymph regions or structures on both sides of the diaphragm
IV
Involvement of extranodal site(s) beyond that designated E
FOR ALL STAGES
A
No symptoms
B
Fever (>38° C [100.4° F]), drenching sweats, weight loss (10% body weight over 6 months)
FOR STAGES I TO III
E
Involvement of a single extranodal site contiguous or proximal to known nodal site
COTSWOLD MODIFICATIONS
(i)
Suffix X to designate bulky disease as more than one third widening of the mediastinum or >10-cm maximum dimension of nodal mass
(ii)
The number of anatomic regions involved should be indicated by a subscript (e.g., 113).
(iii)
Stage III may be subdivided into:
III1, with or without splenic, hilar, celiac, or portal nodes
III2, with para-aortic, iliac, mesenteric nodes
(iv)
Staging should be identified as clinical stage or pathological stage.
(v)
A new category of response to therapy, unconfirmed/uncertain complete remission should be introduced because of the persistent radiologic abnormalities of uncertain significance.
Prognostic Factors for Lymphoma
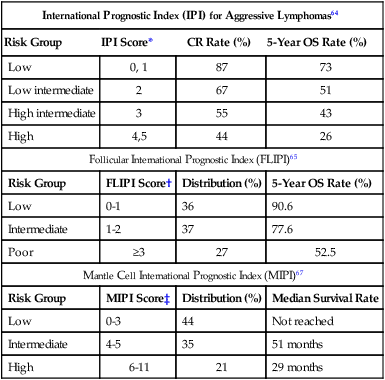
International Prognostic Index (IPI) for Aggressive Lymphomas64
Risk Group
IPI Score*
CR Rate (%)
5-Year OS Rate (%)
Low
0, 1
87
73
Low intermediate
2
67
51
High intermediate
3
55
43
High
4,5
44
26
Follicular International Prognostic Index (FLIPI)65
Risk Group
FLIPI Score†
Distribution (%)
5-Year OS Rate (%)
Low
0-1
36
90.6
Intermediate
1-2
37
77.6
Poor
≥3
27
52.5
Mantle Cell International Prognostic Index (MIPI)67
Risk Group
MIPI Score‡
Distribution (%)
Median Survival Rate
Low
0-3
44
Not reached
Intermediate
4-5
35
51 months
High
6-11
21
29 months
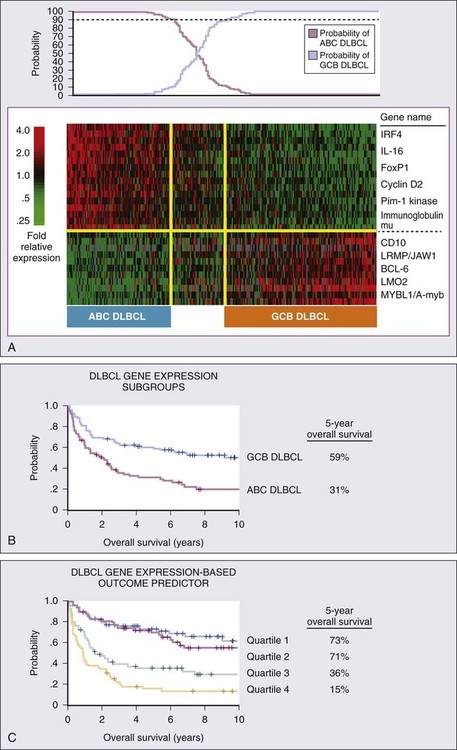
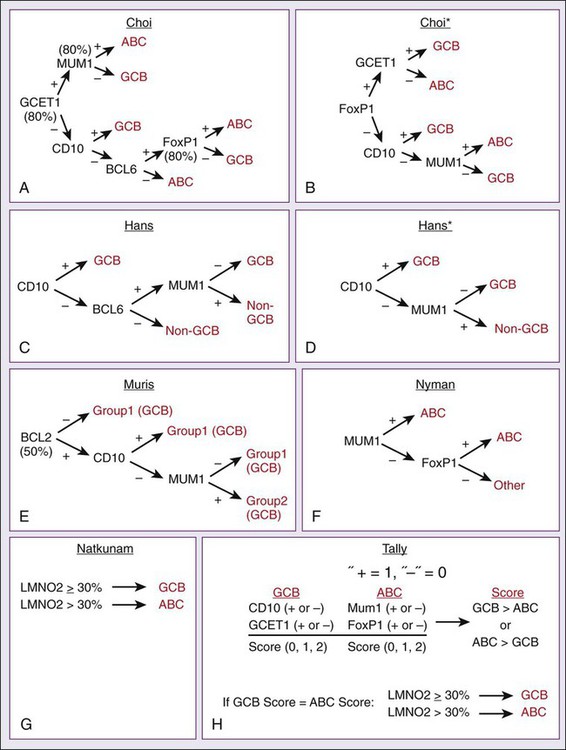
Response Assessment
Oncohema Key
Fastest Oncology & Hematology Insight Engine