With the implementation of genomic technologies into clinical practice, we have examples of the predictive benefit of targeted therapy for oncogene-addicted cancer and identified molecular dependencies in non–small cell lung cancer. The clinical success of tyrosine kinase inhibitors against epidermal growth factor receptor and anaplastic lymphoma kinase activation has shifted treatment emphasize the separation of subsets of lung cancer and genotype-directed therapy. Advances have validated oncogenic driver genes and led to the development of targeted agents. This review highlights treatment options, including clinical trials for ROS1 rearrangement, RET fusions, NTRK1 fusions, MET exon skipping, BRAF mutations, and KRAS mutations.
Key points
- •
With the implementation of genomic technologies into clinical practice, we have examples of the benefit of targeted therapy for oncogene-addicted cancer and identified unique molecular dependencies in non–small cell lung cancer.
- •
The clinical success of tyrosine kinase inhibitors against epidermal growth factor receptor and anaplastic lymphoma kinase activation has shifted treatment to emphasize the genomically defined subsets of lung cancer and genotype-directed therapy.
- •
Continued advances in our understanding of lung cancer biology have validated numerous oncogenic driver genes and have led to the rapid development of targeted agents.
- •
The current available data on ROS1, RET, NTRK, MET, BRAF, and KRAS aberrations in non–small cell lung cancer are presented.
- •
The current state of available trials in this space, mechanism of action of the oncogene, mechanisms of resistance to therapy are presented.
Introduction
Lung cancer remains the leading cause of cancer-related deaths worldwide. Historically, the treatment of advanced epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) wild-type non–small cell lung cancer (NSCLC) has relied on platinum-based chemotherapy with a median overall survival (OS) of approximately 1 year. Within the last several years, immune checkpoint inhibitors have demonstrated efficacy in both squamous and nonsquamous histologies and are now approved agents for second-line treatment, largely based on improved OS.
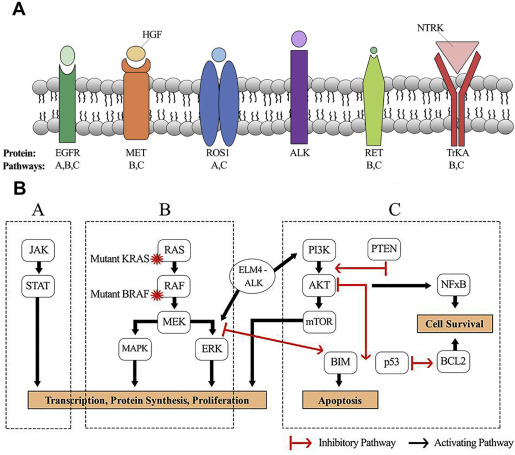
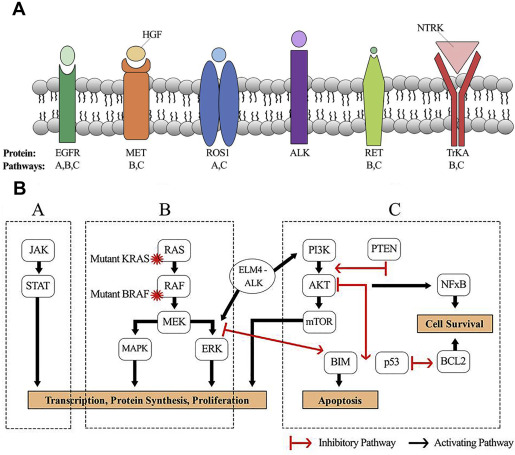
The past decade has seen a paradigm shift in the treatment of NSCLC based on the implementation of broad-based genomics to identify driver mutations and drug development against these targets. Across genotypes, the use of targeted agents for the treatment of patients with EGFR and ALK-mutated NSCLC has improved response rates, time to disease progression, and OS when compared with conventional systemic therapy. More than 60% of patients with lung adenocarcinoma have a presumably mutually exclusive oncogenic driver. This article reviews driver mutations, including ROS1 fusions, RET fusions, NTRK1 fusions, c-MET amplification, exon 14 skipping mutations, BRAF mutations, and KRAS mutations, their downstream signaling ( Table 1 ; Fig. 1 ), and the current clinical trials that are exploring compounds against these pathways.
| Target | Alteration | Frequency (%) |
|---|---|---|
| ROS1 | ROS1 fusion | 2 |
| RET | RET fusion | 1 |
| NTRK1 | NTRK1 fusion | 3.3 |
| c-MET | Amplification | 2–4 |
| Exon 14 skipping mutation | 3–4 | |
| BRAF | V600E mutation | 1–4 |
| KRAS | Mutations in codons 12, 13, and 61 | 15–25 |
Introduction
Lung cancer remains the leading cause of cancer-related deaths worldwide. Historically, the treatment of advanced epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) wild-type non–small cell lung cancer (NSCLC) has relied on platinum-based chemotherapy with a median overall survival (OS) of approximately 1 year. Within the last several years, immune checkpoint inhibitors have demonstrated efficacy in both squamous and nonsquamous histologies and are now approved agents for second-line treatment, largely based on improved OS.
The past decade has seen a paradigm shift in the treatment of NSCLC based on the implementation of broad-based genomics to identify driver mutations and drug development against these targets. Across genotypes, the use of targeted agents for the treatment of patients with EGFR and ALK-mutated NSCLC has improved response rates, time to disease progression, and OS when compared with conventional systemic therapy. More than 60% of patients with lung adenocarcinoma have a presumably mutually exclusive oncogenic driver. This article reviews driver mutations, including ROS1 fusions, RET fusions, NTRK1 fusions, c-MET amplification, exon 14 skipping mutations, BRAF mutations, and KRAS mutations, their downstream signaling ( Table 1 ; Fig. 1 ), and the current clinical trials that are exploring compounds against these pathways.
| Target | Alteration | Frequency (%) |
|---|---|---|
| ROS1 | ROS1 fusion | 2 |
| RET | RET fusion | 1 |
| NTRK1 | NTRK1 fusion | 3.3 |
| c-MET | Amplification | 2–4 |
| Exon 14 skipping mutation | 3–4 | |
| BRAF | V600E mutation | 1–4 |
| KRAS | Mutations in codons 12, 13, and 61 | 15–25 |
The oncogenic ROS1 fusion
ROS1 is a receptor tyrosine kinase (RTK) that belongs to the same insulin receptor superfamily as ALK. The function of ROS1 remains largely unknown, although some studies have suggested that this oncogene may play a role in epithelial cell differentiation. Further progress in the characterization of ROS1 has been difficult owing to the absence of an identified ligand. Expression of the ROS1 fusion protein results in constitutive kinase activity and activation of cellular pathways involved in cell growth and proliferation. Chromosomal rearrangements involving the ROS1 gene were described originally in glioblastoma cell lines and have since been reported in cholangiocarcinoma and NSCLC.
ROS1 gene fusions are found in approximately 2% of NSCLC and have been associated with a younger age of onset and a nonsmoking history, and seem to be mutually exclusive with other oncogenic driver genes. Such demographic characteristics are similar to the clinical profile of patients with ALK-rearranged NSCLC and may be owing in part to the high level of homology between the kinase domains. Crizotinib, a small molecule multikinase inhibitor originally developed as a MET kinase inhibitor and approved by the US Food and Drug Administration (FDA) for the treatment of ALK-rearranged NSCLC, was shown to have potent inhibitory activity against ROS1. Results from the expansion cohort of the PROFILE 1001 (A Study Of Oral PF-02341066, A c-Met/Hepatocyte Growth Factor Tyrosine Kinase Inhibitor, In Patients With Advanced Cancer) trial showed that 72% of the participants experienced a complete or partial response in tumor burden, an effect that lead to a median duration of response of 17.6 months. These findings led to recent FDA approval of crizotinib for patients with ROS1-rearranged NSCLC. Other trials with crizotinib are ongoing ( Table 2 ). Ceritinib, ASP3026, and AP26113 exhibit activity against ROS1 tyrosine kinase, whereas alectinib does not. ROS1-specific agents have not yet entered clinical trials.
| Drug Class and Target | Investigational Agent | Trial ID Number | Phase |
|---|---|---|---|
| ROS1 | Lorlatinib (PF-06463922) | NCT01970865 | I/II |
| Ceritinib (LDK378, SIGNATURE study) | NCT02186821 | II | |
| Cabozantinib | NCT01639508 | II | |
| Entrectinib (RXDX-101, STARTRK-1 study) | NCT02097810 | I | |
| Entrectinib (RXDX-101, STARTRK-2 study) | NCT02568267 | II | |
| Crizotinib (METROS study – Pretreated, metastatic) | NCT02499614 | II | |
| Crizotinib (EUCROSS study – First line Europe) | NCT02183870 | II | |
| Crizotinib (AcSè study – Safety Study) | NCT02034981 | II |
Parallel to experiences seen with EGFR and ALK inhibition, acquired resistance to targeted therapy remains inevitable and has also been described in ROS1-rearranged patients. The secondary G2032R mutation seems to be a dominant resistance mutation and responsible for crizotinib resistance through impaired drug binding. Several second-generation ALK inhibitors that were originally developed to address ALK resistance mutations have failed to inhibit crizotinib-resistant ROS1. Multikinase inhibitors with exquisite inhibition potency when compared with crizotinib are currently being investigated for sensitivity to G2032R mutants and include lorlatinib, entrectinib, and ceritinib (see Table 2 ). Lorlatinib, a selective ALK and ROS1 inhibitor noted to have nanomolar potency, was well-tolerated and demonstrated durable clinical responses in an ongoing phase I study ( NCT01970865 ).
Reactivation of key downstream signaling pathways is another mechanism underlying resistance to targeted therapeutics. Increased EGFR phosphorylation and pathway activation have been reported in crizotinib-resistant NSCLC tumor samples and cell lines. RAS mutations have also been shown to mediate crizotinib resistance in ROS1-rearranged patients. These observations suggest that activation of bypass signaling from the original oncogenic driver to other known drivers. Preclinical studies support combinatorial treatment strategies to overcome acquired resistance mechanisms.
The oncogenic RET fusion
The RET gene encodes a RET family RTK for members of the glial cell line-derived neurotrophic factor family of ligands. RET protein dimerization results in autophosphorylation to activate multiple signal transduction pathways involved in cell motility, proliferation, differentiation, and neuronal nagivation. RET gene rearrangements and point mutations lead to gain of function signaling and subsequent tumorigenesis. Germline and somatic mutations in the RET oncogene are best known to cause the multiple endocrine neoplasia type 2 syndrome (MEN2), nearly all inherited cases of medullary thyroid cancer, and up to 50% of sporadic thyroid cancer cases.
RET fusions were initially identified in lung cancer in 2012 and have since been reported to occur in 1% of NSCLC with adenocarcinoma as the predominant histology. Studies have shown that RET fusions may be associated with a never smoker status and are mutually exclusive with known driver oncogenes. The KIF5B-RET fusion is the most common alteration and accounts for approximately 90% of the rearrangements with fusion partners CCDC6 and NCOA4 representing the remaining 10%.
Although RET-specific inhibitors have not yet been developed, there are numerous clinical trials looking at the efficacy of multikinase inhibitors for advanced NSCLC harboring RET rearrangements ( Table 3 ). Treatment with sunitinib and vandetanib has been reported to induce prolonged antitumor activity in several case reports of patients with RET fusion–positive lung cancer. The drug vandetanib is FDA approved for the treatment of medullary thyroid cancer and showed early results in the phase II LURET (Vandetanib in Advanced NSCLC With RET Rearrangement) study of 17 NSCLC patients with an objective response rate (ORR) of 53% and disease control rate (DCR) of 88% and a median progression-free survival (PFS) of 4.7 months. Treatment response was much higher in patients with the CCDC6-RET fusion subtype with an ORR and median PFS of 83% and 8.3 months, respectively. The ORR and median PFS was only 20% and 2.9 months for those with KIF5B-RET fusion. Results from a different phase II trial of vandetanib in 18 patients with RET-rearranged lung cancer did not show the robust responses seen in the LURET study ( NCT01823068 ). Three patients were reported to have a partial remission, 8 were reported to have stable disease, and the remaining 7 patients had disease progression. Interim results of a phase II trial of cabozantinib in patients with RET -rearranged lung cancers presented last year reported an ORR of 38% ( NCT01639508 ). These findings have suggested that a proportion of tumors may harbor intrinsic resistance mechanisms to RET inhibition or there may be different efficacy based on different fusion partners. Recent molecular studies performed on pre-treatment and post-treatment tumor biopsies from patients treated with cabozantinib have identified MDM2 amplification as a potential mechanism of primary or acquired resistance. Cell lines and xenografts with acquired MDM2 amplification have demonstrated significant tumor growth suppression in response to MDM2 inhibition.
| Drug Class and Target | Investigational Agent | Trial ID Number | Phase |
|---|---|---|---|
| RET | Lenvatinib (E7080) | NCT01877083 | II |
| Apatinib | NCT02540824 | II | |
| Vandetanib (LURET study) | NCT01823068 | II | |
| Ponatinib | NCT01813734 | II | |
| Cabozantinib | NCT01639508 | II | |
| Pazopanib | NCT02193152 | 0 | |
| Nintedanib | NCT02299141 | 0 | |
| Ponatinib | NCT01935336 | II | |
| Vandetanib + Everolimus | NCT01582191 | I |
The rapid emergence of resistance to RET inhibition underscores the need for combinatorial treatment strategies. In vitro studies after treatment with vandetanib, everolimus, or in combination were performed on 2 cell lines expressing the CCDC6-RET fusion subtype. Vandetanib was shown to abrogate signaling through the MAPK pathway with effects on effectors of the mammalian target of rapamycin (MTOR). A phase I study of vandetanib in combination with everolimus was shown to be safe and tolerable with significant activity in RET-rearranged lung cancers ( NCT01582191 ). Of the 6 patients who harbor RET fusions by next-generation sequencing, 5 achieved a partial response, including 1 patient who progressed on cabozantinib. Significant antitumor activity in the central nervous system was seen with the combination.
Analysis of a global registry of RET -rearranged NSCLC included 41 patients who received RET inhibitor therapy off protocol and reported a median PFS of 2.9 months and an OS of 6.8 months with distributions of patients including the major RET fusion partners. For 35 patients who had serial imaging evaluated by Response Evaluation Criteria In Solid Tumors (RECIST) criteria, the ORR was 23% and DCR was 57%. This proportion is lower than that observed with targeted therapy for EGFR-mutant and ALK-rearranged NSCLC. These results emphasize the need for an improved understanding of the intrinsic tumor biology in this genotype.
The oncogenic NTRK1 fusion
NTRK1 is an RTK that belongs to the tropomyosin-related kinases superfamily. Tropomyosin-related kinase signaling is normally involved in the regulation of neural development and maintenance of neural networks. Ligand binding to NTRKs leads to the activation of multiple signaling pathways through mitogen-activated protein kinase (MAPK), phospholipase C-γ, and phospoinositide 3-kinase to mediate cell differentiation and survival. The NTRK1 gene encodes the high-affinity nerve growth factor receptor (TrkA). Fusions involving the NTRK1 gene result in expression of TrkA fusion proteins with constitutive activity or overexpressed kinase function, hence conferring oncogenic potential. NTRK1 gene rearrangements were first reported in colorectal cancer several decades ago and have now been reported in other solid tumor malignancies, including sarcomas.
Oncogenic NTRK1 fusions have been identified in 3.3% of lung cancer cases with adenocarcinoma histology that is negative for other common driver mutations. Two different gene fusions have been previously described and are characterized by rearrangements between the myosin phosphatase Rho-interacting protein gene ( MPRIP-NTRK1) and CD74 gene ( CD74-NTRK1 ). Both in-frame gene fusion events have been detected in female never smokers. Additional NTRK1 gene rearrangements have been reported in other studies as well. These mutations play an important role in tumorigenesis and drug development has moved quickly against these therapeutic targets.
Numerous phase I trials are in progress with inhibitors targeted toward specific kinases that are active against NTRK ( Table 4 ). Entrectinib is a highly potent, selective, orally bioavailable small molecule inhibitor of TrkA, TrkB, TrkC, ROS1, and ALK that has achieved rapid and durable responses across a wide range of advanced malignancies. A phase I dose escalation study of entrectinib (STARTRK-1 [Study of Oral RXDX-101 in Adult Patients With Locally Advanced or Metastatic Cancer Targeting NTRK1, NTRK2, NTRK3, ROS1, or ALK Molecular Alterations] study) demonstrated significant antitumor activity in a patient with metastatic NSCLC harboring a SQSTM1-NTRK1 gene rearrangement with RECIST-defined partial response and complete response of all brain metastases ( NCT02097810 ). The patient has been on study for more than 12 months with no evidence of progressive disease. Entrectinib was granted FDA Orphan Drug Designation for the treatment of TrkA-, TrkB-, and TrkC-positive NSCLC and colorectal cancers.
| Drug Class and Target | Investigational Agent | Trial ID Number | Phase |
|---|---|---|---|
| NTRK1 | LOXO-101 | NCT02122913 | I |
| LOXO-101 (NAVIGATE study) | NCT02576431 | II | |
| Entrectinib (RXDX-101, STARTRK-1 study) | NCT02097810 | I | |
| Entrectinib (RXDX-101, STARTRK-2 study) | NCT02568267 | II | |
| Cabozantinib | NCT01639508 | II | |
| Sitravatinib (MGCD516) | NCT02219711 | I | |
| TSR-011 | NCT02048488 | I/IIa | |
| Altiratinib (DCC-2701) | NCT02228811 | I | |
| DS-6051b | NCT02279433 | I | |
| PLX7486 | NCT01804530 | I |
A number of phase II trials with entrectinib and other novel NTRK inhibitors are underway with compounds including LOXO-101, entrectinib, and cabozantinib (see Table 4 ). Two phase II basket studies (NAVIGATE LOXO-101 [Study of LOXO-101 in Subjects With NTRK Fusion Positive Solid Tumors] and STARTRK-2 [Basket Study of Entrectinib (RXDX-101) for the Treatment of Patients With Solid Tumors Harboring NTRK1/2/3, ROS1, or ALK Gene Rearrangements (Fusions)] entrectinib) are currently recruiting patients who have advanced solid tumors harboring NTRK fusions ( NCT02576431 and NCT02568267 ). Efficacy data are eagerly awaited to determine whether results seen from the phase I studies can be confirmed in a larger cohort of patients. These findings highlight the clinical utility of molecular testing to identify patients with a subset of lung cancers sensitive to NTRK inhibition. Other agents in earlier stages of development include TSR-011, sitravatinib, altiratinib, DS-6051b, and PLX7486 (see Table 4 ).
Amplification and mutation in c-MET
c-MET is an RTK in the MET/RON family whose only known ligand is hepatocyte growth factor. Ligand binding promotes MET dimerization and phosphorylation to recruit signaling protein complexes including SRC, GRB2, SHC, and the p85 regulatory unit of the phospoinositide 3-kinase, which are required for the activation of several downstream signaling cascades. The presence of hepatocyte growth factor also leads to MET receptor internalization and downregulation whereby they are either recycled to the cell surface or degraded within lysosomes. Degradation of the MET receptor depends on ubiquitination by the Cbl family of ubiquitin–protein ligases. A receptor mutation at tyrosine 1003 (Y1003) abrogates MET ubiquitination and results in elevated MET protein levels and increased protein stability. This specific uncoupling leads to oncogenic activation by sustained activation of the RAS–MAPK pathway.
Abnormal c-MET signaling has been implicated in a wide variety of malignances, including renal cell carcinoma and lung cancer. Germline mutations in MET occur in all cases of hereditary papillary renal cell carcinoma and are also found in 10% to 15% of sporadic cases. Two main mechanisms of aberrant MET activation have been reported in lung cancer, including MET gene amplification and MET exon 14 skip mutations. MET gene amplification has been reported to occur in 2% to 4% of NSCLC. MET exon 14 skipping mutations have been found in 3% to 4% of lung adenocarcinoma with and without gene amplification. Increased MET expression has been associated with aggressive tumor biology and is a negative prognostic factor. The high frequency of MET overexpression in NSCLC has generated significant clinical interest in this pathway for potential druggable targets.
Acquired MET amplification has been associated with secondary resistance across anti-EGFR tyrosine kinase inhibitors. This phenomenon may occur in up to 20% of treated patients and has also been reported to cause resistance to selective ALK inhibition. Clinical trials investigating treatment with dual EGFR and MET inhibition are underway using compounds including crizotinib, glesatinib, capmatinib, emibetuzumab, and cabozantinib ( Table 5 ). Onartuzumab, an anti-MET monoclonal antibody, did not improve efficacy endpoints when combined with erlotinib in a phase III study and was stopped early for futility. More recently, results from a phase II trial combining emibetuzumab (LY2875358), a humanized immunoglobulin G4 bivalent monoclonal MET antibody blocking ligand-dependent and ligand-independent hepatocyte growth factor/MET signaling, with erlotinib found that dual inhibition was unable to overcome acquired resistance to erlotinib in patients with advanced NSCLC. There are, however, subsets of patients with response and ongoing work is focused on defining biomarkers and the mechanisms of those patients with response.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


