This review describes developments in understanding normal mast cell function and genetic changes that predispose to malignant transformation to mastocytosis. Most mastocytosis cases are associated with somatically acquired activating mutations in the KIT receptor. The role these mutations play in the development of mastocytosis is discussed. Mastocytosis is classified into cutaneous mastocytosis and systemic mastocytosis. The classification of mastocytosis and clinical presentation of each variant is detailed in this report. Progress has been made in developing drugs that target the wild-type and mutated KIT receptor, and these and other new therapeutic strategies for the treatment of mastocytosis are reviewed.
Key Points
- •
Mast cells (MCs) play a role in both innate and adaptive immunity and are an important component of the host response to pathogens and environmental toxins.
- •
The principal growth factor for MCs is Stem Cell Factor (SCF), which controls differentiation and survival by binding to the KIT receptor tyrosine kinase.
- •
Somatically acquired mutations in the KIT receptor are central to the pathogenesis of mastocytosis, a myeloid malignancy characterized by the accumulation of malignant MCs.
- •
Drugs which target KIT have efficacy in the treatment of mastocytosis; however the common activating mutation is resistant to most KIT inhibitors. Identification of other upregulated signaling pathways will provide additional targets for future therapies.
Introduction
Mast cells (MCs) are derived from bone marrow progenitor cells, which migrate via the peripheral blood to reside in tissues where they are found closely associated with blood vessels and nerve fibers. In particular, MCs are found in high numbers in the skin and mucosal surfaces where it has been recognized that they play an important role in both innate and adaptive immune responses to pathogens. Mastocytosis (MIM 154 800) is a rare myeloid malignancy that is characterized by the abnormal accumulation of malignant MCs in one or several body tissues. Most cases are sporadic and associated with somatically acquired activating mutations in the KIT oncogene. However, a small number of familial cases have been described; the occurrence of mastocytosis with concordant clinical features in sets of monozygotic twins indicates that a subset of cases occur as a consequence of an inherited genetic predisposition. The disease is divided into 2 main categories: cutaneous mastocytosis (CM), in which the MC infiltrate is confined to one or more lesions on the skin, and systemic mastocytosis (SM), which is characterized by MC infiltration of at least one extracutaneous organ with or without evidence of skin involvement. In addition to symptoms and signs attributable to specific organ involvement, most patients report symptoms related to the release of MC mediators that include histamine, various cytokines, and eicosanoid lipid mediators. Although a significant proportion of pediatric cutaneous cases spontaneously regress before adolescence, most MC neoplasms occur in association with a mutation in the catalytic site of KIT, which is refractory to currently available treatments.
Introduction
Mast cells (MCs) are derived from bone marrow progenitor cells, which migrate via the peripheral blood to reside in tissues where they are found closely associated with blood vessels and nerve fibers. In particular, MCs are found in high numbers in the skin and mucosal surfaces where it has been recognized that they play an important role in both innate and adaptive immune responses to pathogens. Mastocytosis (MIM 154 800) is a rare myeloid malignancy that is characterized by the abnormal accumulation of malignant MCs in one or several body tissues. Most cases are sporadic and associated with somatically acquired activating mutations in the KIT oncogene. However, a small number of familial cases have been described; the occurrence of mastocytosis with concordant clinical features in sets of monozygotic twins indicates that a subset of cases occur as a consequence of an inherited genetic predisposition. The disease is divided into 2 main categories: cutaneous mastocytosis (CM), in which the MC infiltrate is confined to one or more lesions on the skin, and systemic mastocytosis (SM), which is characterized by MC infiltration of at least one extracutaneous organ with or without evidence of skin involvement. In addition to symptoms and signs attributable to specific organ involvement, most patients report symptoms related to the release of MC mediators that include histamine, various cytokines, and eicosanoid lipid mediators. Although a significant proportion of pediatric cutaneous cases spontaneously regress before adolescence, most MC neoplasms occur in association with a mutation in the catalytic site of KIT, which is refractory to currently available treatments.
Biologic and molecular aspects
MC Function
MCs are widely distributed throughout the body but are particularly found near surfaces exposed to the environment, including the skin, airways, gastrointestinal tract, and genitourinary tract. As a consequence, in conjunction with dendritic cells (DCs) and macrophages, MCs are among the first cells to encounter and help initiate an immune response to pathogens and environmental toxins. MCs can express a range of receptors that detect products generated at sites of infection, such as pathogen-specific antibodies that bind to MC Fc receptors, and they directly recognize pathogens through pattern recognition receptors, including the Toll-like receptors (TLRs) TLR4 and TLR2, which are activated in response to pathogen-associated molecular patterns (PAMPs). MCs undergo degranulation in response to exogenous venoms and other antigens that are injected into the skin, which recruits inflammatory cells to the sites of insect bites and may have significance to the immune response against insect-borne pathogens. Finally, several host endogenous peptides and substances generated at sites of inflammation can activate MCs. These substances include neurotensin; substance P; endothelin; and complement components, particularly C5a, which can induce MC degranulation. Following activation, MC populations expand by recruitment and differentiation of circulating MC progenitors. Both innate and acquired immune responses, in particular T H 2-type responses, are associated with increased numbers of MCs at sites of inflammation.
MCs share many functions with macrophages, which include host response to damage-associated molecular pattern molecules and PAMPs, antigen presentation, and phagocytosis. In a 2-wave process, MCs are able to secrete a large number of preformed and newly synthesized products that can amplify or suppress both the innate and adaptive immune responses. Within seconds of MC activation, preformed mediators, including proteases, histamine, and tumor necrosis factor (TNF), which are stored in cytoplasmic granules, are released at the site of inflammation. This release is followed by the de novo production of eicosanoids, prostaglandins, and leukotrienes, which are released within minutes, and cytokines, including interleukin 4 (IL-4), which are released within hours.
MCs are categorized into 2 groups based on the protease content of their vesicles as either predominantly tryptase (MC T cells) or, less commonly, tryptase and chymase (MC TC cells). Because these proteases have different substrate specificities, factors that regulate the required MC function will determine the predominant MC subtype. MC T cells are the predominant type found in the respiratory and gastrointestinal mucosa and are increased during inflammation, whereas MC TC cells are found in the connective tissue of the dermis and gastrointestinal tract.
Recruitment of immune cells to sites of bacterial infection during the innate immune response is facilitated by MC release of histamine, TNF, vascular endothelial growth factor, and proteases, which contribute to increased local vascular permeability and edema. In addition, chemokines produced by MCs attract other innate immune cells, including neutrophils, eosinophils, and natural killer cells, to inflamed tissues. This process may operate during viral infections and is probably stimulated by double-stranded RNA. In addition, some of the products released by MCs are directly bactericidal, including reactive oxygen species and cathelicidins, which assist in killing group A streptococci.
Following stimulation, MCs participate in modulating the adaptive immune response. At the site of infection, MCs promote the influx of monocyte-derived DCs and, in response to bacterial peptidoglycan and gram-negative bacteria, activate local Langerhans cells. In addition to recruiting antigen-presenting cells (APCs), there is in vivo evidence that MCs themselves function as APCs for major histocompatibility complex class I–restricted CD8 + T cells. Depending on the stimulus, MC function can be altered by the amount and pattern of expression of cell surface receptors, which enhance or suppress particular immune responses. In response to parasitic infections and in allergic disorders, high concentrations of circulating immunoglobulin E (IgE) induce increased surface expression of high-affinity receptor for IgE (FcϵRI) by tissue MCs, which enhance IgE-dependent effector functions. However, for allergens, this response is pathologic; FcϵRI-mediated MC activation may play a role in the pathogenesis of autoimmune diseases, including bullous pemphigoid, rheumatoid arthritis, and multiple sclerosis. There is also evidence that MCs may reduce the duration and response of immune responses either directly through the secretion of IL-10 or indirectly via CD4 + CD25 + FoxP3 + regulatory T-cell–dependent peripheral tolerance.
MC Differentiation
Paul Ehrlich first described MCs in 1878; for the subsequent 100 years, it was thought that they were derived from mesenchymal cells and constituted a component of connective tissue. However, in the late 1970s and early 1990s, it was shown that MCs developed from multipotent hematopoietic stem cells, which are released into the blood as MC precursors and complete their differentiation within connective tissue. The principal growth factor for MCs is stem cell factor (SCF) or KIT ligand ( Fig. 1 ), which controls differentiation and survival by binding to the KIT protein tyrosine kinase (CD117). Proliferation of MC progenitors is supported by the presence of IL-6 and IL-11, which both share a common signal-transducing component. In addition to its role as a growth factor, SCF is a potent chemoattractant that induces MC motility and accumulation at sites of infection or inflammation. Both MC subtypes, MC T and MC TC , develop from a common precursor cell; MC T cells maintained in SCF-containing media can be induced to express chymase and upregulate FcϵRI after exposure to IL-4. In addition, in vitro studies have shown SCF in combination with IL-6, IL-1β, transforming growth factor beta 1, or lipopolysaccharide can induce MC T cells to increase chymase expression.
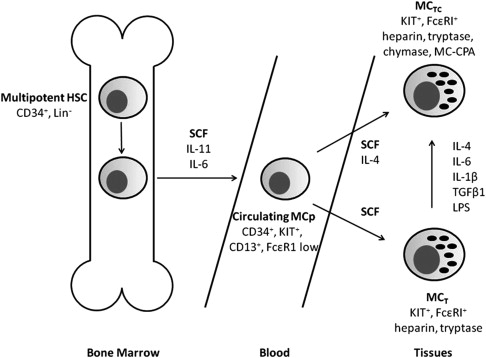
Following SCF binding, KIT is activated by dimerization and autophosphorylation of tyrosine residues. This activation results in the activation of several signaling pathways, including phosphoinositide 3-kinase, phospholipase C, protein kinase C, the Ras–mitogen-activated protein kinase cascade, and the Jak-Stat pathway, which are all critical for MC development. The presence in tissues of MCs requires an intact SCF/KIT pathway such that mice with loss-of-function mutations in the Kit gene, which do not express functional Kit receptors, are devoid of MCs. Furthermore, in this murine model, normal MC development and survival can be rescued by retroviral gene transfer of normal wild-type Kit. In addition to playing a central role in migration and differentiation, SCF is involved in the regulation of MC survival by preventing apoptosis. KIT signaling following the treatment of MCs with SCF results in increased levels of the Bcl-2 family proteins, Bcl-2 and Bcl-X L , which promote MC survival. Furthermore, the levels of the proapoptotic Bcl-2 family members, Bad and Bim, are decreased, which in combination with Bcl-2 overexpression prevents MC apoptosis, particularly when associated with growth factor deprivation.
Pathogenesis of Mastocytosis
Considering the crucial role of SCF and KIT signaling for MC survival, several investigators screened for mouse Kit and human KIT mutations in MC lines and identified several gain-of-function mutations. Moreover, the introduction of an activating murine Kit mutation into MCs was found to be sufficient to induce ligand-independent growth, differentiation, and tumorigenicity in vivo.
KIT belongs to the type III transmembrane receptor tyrosine kinase subfamily and contains extracellular, transmembrane, and juxtamembrane domains and an enzymatic pocket domain, which consists of a kinase insert, an adenosine triphosphate–binding site, and an activation loop. Mutations in the human KIT protein at codon 816, which is located within the enzymatic pocket, induce ligand-independent phosphorylation and activation, whereas mutations in the juxtamembrane domain cause disruptions of the regulation of phosphorylation and kinase activity of KIT and are labeled “regulatory type mutations.” Based on data using cell line models, investigators screened human mastocytosis cases for KIT mutations and found that aspartic acid to valine at codon 816 (KIT D816V) was the most common activating mutation. This mutated KIT is found in more than 90% of patients with SM, including both indolent and aggressive subgroups. In contrast, although activating KIT mutations are found in most pediatric patients with CM, D816V is present in only one-third of patients.
Although mutations in KIT clearly play a role in the development of mastocytosis, it has not been shown that the presence of KIT D816V in isolation is sufficient to induce malignant transformation of MCs. The murine equivalent of KIT D816V, the Kit D814V mutation, has transforming activity in vitro and in vivo in tumorigenicity assays; however, these effects have not been observed for the human KIT D816V mutation. Expression of KIT D816V in a murine IL-3–dependent cell line has been shown to induce cluster formation and MC differentiation antigens, which was not observed in wild-type KIT. However, cells were not converted to factor-independent proliferation. Therefore, it has been proposed that D816V in isolation may be sufficient to cause indolent mastocytosis, but additional somatic genetic changes are necessary to induce aggressive mastocytosis. There is evidence that signaling pathways independent of KIT, including phosphorylation of the nonreceptor protein kinases Lyn and Btk, and somatic mutations in the putative tumor suppressor gene TET2 provide additional mechanisms for malignant transformation of MCs.
Rare familial mastocytosis cases with germline mutations of KIT have been reported, which include activating mutations in the extracellular and juxtamembrane domains of KIT. It is important to identify these cases, which (in contrast to D816V cases) may show response to specific tyrosine kinase inhibitors.
In CM, both activating and inactivating KIT mutations have been detected from MCs in skin samples but rarely from MCs in blood or bone marrow. The reported prevalence in skin samples from pediatric CM cases of codon 816 KIT mutations, which includes KIT D816V, has varied from 0% to 83% ; initially, it was proposed that the presence of these activating mutations predicted progression to SM. However, this finding has not been replicated in later studies that showed patients with codon 816 mutations, including between 66% and 70% of D816V cases, failed to segregate with progressive or persistent disease.
Classification of mastocytosis
Diseases that result from the pathologic accumulation of MCs in various tissues are classified into cutaneous (CM) and systemic forms (SM), which are further subclassified into several variants and subvariants ( Table 1 ).
| Variant | Diagnostic Criteria | Subvariants | Prognosis |
|---|---|---|---|
| CM | Typical clinical findings & histologic proof of abnormal MC dermal infiltrate; No systemic involvement (most patients are children) | Urticaria pigmentosa/maculopapular CM Diffuse CM Solitary mastocytoma of the skin | Good |
| Indolent SM (ISM) | No impaired organ function; MC burden is low and skin lesions are usually present |
| >16-y expected median survival and not significantly different from age- and sex-matched controls |
| Systemic mastocytosis with an associated clonal hematologic non-MC lineage disease (SM-AHNMD) | Meets criteria for SM and WHO criteria for an associated, clonal hematological non-MC lineage disorder | SM-AML SM-MDS SM-MPN SM-CEL SM-CMML SM-NHL SM-myeloma | 2-y expected median survival, dependent on underlying AHNMD |
| Aggressive systemic mastocytosis |
|
| 3.5-y expected median survival |
| MC leukemia | Meets criteria for SM; bone marrow aspirate shows >20% atypical, immature MCs and peripheral blood ≥10% MCs |
| 2-mo expected median survival |
| MCS | Unifocal malignant tumor destroying the soft tissue; no evidence of SM | Very poor; transformation to MCL after a short interval | |
| Extracutaneous mastocytoma | Extremely rare benign tumor; nondestructive growth pattern and low-grade cytology | Good |
The clinical phenotypes of mastocytosis are heterogeneous; variants are recognized mainly by the distribution of organ involvement and clinical course, which can vary from the presence of benign cutaneous lesions that spontaneously regress to highly aggressive disease with short survival. Mastocytosis is a clonal malignancy; the KIT D816V somatic mutation can be detected in myeloid, non-MC lineage cells. Consequently, mastocytosis is classified as a myeloproliferative neoplasm in the World Health Organization’s (WHO) classification of tumors. Following this classification, the diagnosis of mastocytosis requires the presence of typical clinical findings and a histologic demonstration of an abnormal MC infiltrate in tissue biopsies ( Box 1 ). In cases of CM, there must be no evidence of systemic disease, such as elevated serum tryptase level or organomegaly. To establish a diagnosis of SM, patients must fulfill at least one major and one minor SM criterion or 3 minor criteria (see Box 1 ).
CM
There is demonstration of typical skin lesions, which includes urticaria pigmentosa/maculopapular CM, diffuse CM, or solitary mastocytoma, and histologic evidence of an abnormal MC infiltrate in the dermis. There must be insufficient criteria to establish a diagnosis of SM.
SM
The diagnosis of SM can be established if at least one major and one minor or at least 3 minor criteria are fulfilled.
Major criterion
Multifocal dense infiltrates of MCs (>15 MCs in aggregate) are found in tissue sections of bone marrow or other extracutaneous organs.
Minor criteria
Greater than 25% of MCs in bone marrow or extracutaneous organs are spindle-shaped or have abnormal morphology or, in bone marrow smears, more than 25% are immature or atypical.
There is detection of an activating KIT mutation at codon 816 in blood, bone marrow, or another extracutaneous organs.
MCs in bone marrow, blood, or other extracutaneous organs express CD2 and/or CD25.
Serum total tryptase is persistently more than 20 ng/mL (invalid in patients who have associated clonal hematologic non-MC lineage disease).
Following diagnosis, SM is staged as indolent, smoldering, or aggressive based on the presence or absence of B and C findings ( Box 2 ). B findings indicate a high MC burden and/or expansion of the genetic defect into other myeloid lineages, whereas C findings result from impaired organ function caused by MC infiltration, which requires confirmation by biopsy in most cases. The criteria for smoldering mastocytosis are fulfilled if 2 or more B findings are present but there is no evidence of impaired organ function. If one or more C findings are present, then patients are classified as either aggressive SM (ASM) or MC leukemia (MCL), and treatment using cytoreductive therapy is considered. Investigations are performed to detect the presence of an additional hematologic non-MC neoplasm (SM-associated clonal hematologic non–mast cell lineage disease [AHNMD]), which will have prognostic implications and specific treatment requirements.
B findings
Bone marrow biopsy showing more than 30% MCs and/or serum total tryptase levels more than 200 ng/mL
Bone marrow biopsy showing dysplasia or myeloproliferation in non-MC lineages but without substantial cytopenias or WHO criteria for a myelodysplastic syndrome or myeloproliferative neoplasm
Palpable hepatomegaly, splenomegaly, or lymphadenopathy (on computed tomography or ultrasound >2 cm) without impaired organ function
C findings
Bone marrow dysfunction with one or more cytopenias: absolute neutrophil count less than 1 × 10 9 /l, or hemoglobin less than 100 g/L, or platelets less than 100 × 10 9 /l
Palpable hepatomegaly with impaired liver function, ascites and/or portal hypertension
Palpable splenomegaly with hypersplenism
Skeletal lesions with large osteolytic lesions and/or osteoporosis causing pathologic fracture
Malabsorption with hypoalbuminemia and weight loss caused by gastrointestinal MC infiltration
Epidemiology
Mastocytosis occurs in all ethnic groups and may present at any age. CM is more common in children ; but a second smaller peak of incidence is seen in adults in the third to fourth decade. About 50% of children develop CM before 6 months of age, and the average duration of disease is estimated at 9.4 years. Improvement or resolution of disease is reported in most of these cases. However, in adults, there is a high risk of CM progressing to SM, which should be suspected in adults with urticaria pigmentosa (UP) skin lesions at presentation. In contrast, SM is more common in adults and usually diagnosed after the second decade. CM has a slight male predominance, whereas SM affects men and women equally.
Clinical and laboratory features at presentation
Mastocytosis is comprised of several heterogeneous diseases; consequently, the presenting clinical features are diverse. To a large extent, the clinical manifestations and disease course are related to the age of presentation, which can be classified into pediatric- and adult-onset groups. Mastocytosis in the pediatric group is usually confined to the skin; the most common pattern of disease is UP, which represents 70% to 90% of cases. The clinical course of pediatric CM is variable but tends toward spontaneous resolution before puberty. In contrast, mastocytosis in adults usually presents with symptoms and signs of systemic disease that progress over time. Overall, approximately 80% of patients will have signs of skin involvement. The skin is involved in more than 50% of SM cases, which may predict a more indolent course and is usually not a feature of more aggressive SM variants.
The symptoms of SM are usually grouped into 4 categories: (1) constitutional symptoms, which include fatigue, weight loss, sweats, and fever; (2) skin symptoms; (3) MC mediator-related symptoms; and (4) musculoskeletal symptoms, which include bone, muscle, and joint pain. The signs at diagnosis of SM include splenomegaly and, less commonly, lymphadenopathy and hepatomegaly. SM cases may also have signs and symptoms of bone marrow infiltration and failure, which include recurrent bacterial infections, bruising and bleeding, and symptoms of anemia.
CM
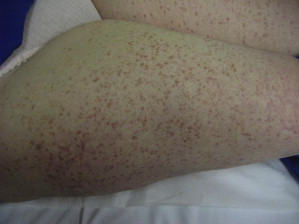
The WHO classification of CM includes 3 subvariants: UP, diffuse CM, and mastocytoma of the skin. UP is the most common form of CM and usually presents with disseminated brown or red macules and papules that spare the face, palms, and soles of the feet ( Fig. 2 ).
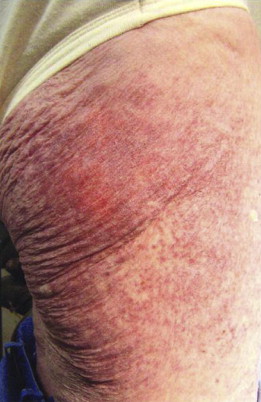
Physical stroking of the lesions causes MC degranulation, which is observed as a wheal and flare reaction and constitutes a positive Darier sign. This urticarial reaction, in combination with intraepidermal accumulation of melanin pigment, explains the descriptive term urticaria pigmentosa ( Fig. 3 ).
Bullous skin lesions can occur in all forms of pediatric CM but are seen more commonly in children less than 3 years of age and in mastocytoma of the skin and diffuse CM subvariants. In general, pediatric CM is usually asymptomatic or minimally symptomatic. However, all forms of CM can present with systemic symptoms because of the local release of MC mediators, which have both local and systemic effects. Skin symptoms may occur spontaneously or be triggered by mechanical or thermal stimuli and include flushing, pruritus, swelling, and dermatographism. Systemic symptoms, including diarrhea, wheezing, and syncope, can accompany pediatric UP but seem to be more common in adult-onset UP and, in one series, were reported in 25% of cases. Based on distinct clinical features, several rare subvariants of UP have been described, which include a plaque form, a nodular form, and a telangiectatic variant (telangiectasia macularis eruptiva perstans).
Mastocytoma of the skin usually presents in the first 3 months of life with 1 to 3 plaques or nodules that are generally larger than 1 cm in diameter. The lesions are brown or orange and are usually located on the extremities but spare the palms and soles of the feet. Bullous lesions, which occur spontaneously or after trauma, can be seen in both mastocytoma of the skin and diffuse CM. Diffuse CM is an extremely rare form of CM, which is characterized by a widespread and heavy skin MC load. Diffuse CM presents in infancy, with erythroderma involving almost the entire body and is often associated with hemorrhage and prominent blister formation. Systemic symptoms are pronounced as a result of the high MC load, although the incidence of systemic involvement by MCs is unknown. In the rare reported cases, acute cutaneous flares of the disease have been associated with significant mortality. However, in those that survive these episodes, spontaneous resolution has usually occurred before 5 years of age.
SM
Indolent SM (ISM) is the most frequently diagnosed subvariant of SM and composed about half of the cases in the largest series reported by Lim and colleagues. Patients with ISM are younger and experience a longer duration of symptoms before diagnosis compared with other SM subvariants. Patients with ISM are also more likely to have skin involvement with UP lesions and report a higher prevalence of MC mediator-related symptoms. The life expectancy of patients with ISM is not significantly different than age- and sex-matched controls ; but those affected can experience a wide variety of symptoms that arise from MC mediator release, which may significantly impact their quality of life. These symptoms can include pruritus, urticaria, angioedema, flushing, bronchoconstriction, abdominal pain, nausea, vomiting, diarrhea, hypotension, and anaphylactoid reactions. Gastric acid secretion is stimulated by histamine, which can cause gastric ulceration and lead to complications, including bleeding and perforation. In addition, it has become increasingly recognized that neuropsychiatric manifestations, particularly the psychological impact of the cosmetic appearance of skin lesions, significantly contribute to disability in mastocytosis.
SM associated with another hematological non-MC disease (SM-AHNMD) and ASM compose 40% and 12%, respectively, of SM cases. In contrast to ISM, ASM and SM-AHNMD are associated with a higher frequency of constitutional symptoms, hepatosplenomegaly, and lymphadenopathy. Patients with SM-AHNMD are generally older and have a shorter survival time compared with ISM, which is usually related to the underlying AHNMD rather than SM. Almost 90% of cases have an associated myeloid neoplasm, with the remainder associated with lymphoma, myeloma, chronic lymphocytic leukemia, and rarely, primary amyloidosis. Signs and symptoms of hematological abnormalities are seen in both ASM and SM-AHNMD. Approximately one-third of patients with SM-AHNMD and one-quarter of patients with ASM are anemic (hemoglobin <10 g/dL) and thrombocytopenic (platelets <100 × 10 9 /L), and 20% to 30% of cases show a prominent eosinophilia (absolute eosinophil count >1500/μL).
MCL is rare and is characterized by circulating MCs and 20% or greater MCs on the bone marrow aspirate smear. Most patients are adults. Cutaneous lesions are typically absent; patients initially present with episodes of mediator-related symptoms and later develop constitutional symptoms, including weight loss and bone pain, and symptoms and signs of organomegaly.
MC sarcoma is extremely rare and characterized by local destructive proliferation of highly atypical MCs, which rapidly progresses to a terminal phase that is indistinguishable from MCL. Reported primary sites of involvement include the larynx, colon, meninges, bone, and skin ; however the bone marrow is usually not initially affected. At first presentation, it is important to differentiate MC sarcoma from extracutaneous mastocytoma, which is a localized benign MC tumor without systemic involvement. Extracutaneous mastocytomas are characterized by low-grade cytology and a nondestructive growth pattern and have been primarily found in the lungs.
Varieties of skeletal changes are associated with SM and differ in frequency depending on the subtype of disease; however, overall, about 50% of patients will have detectable bone involvement. Diffuse osteosclerosis is observed in up to one-third of patients with ASM but is rare in ISM, which is more commonly associated with mixed osteosclerotic and osteolytic lesions. An increased risk of osteopenia and osteoporosis is well described in all forms of SM, with a prevalence between 18% and 31% ; the prevalence of osteoporotic fractures is significantly higher than controls.
Diagnosis
Histology and Immunohistochemistry
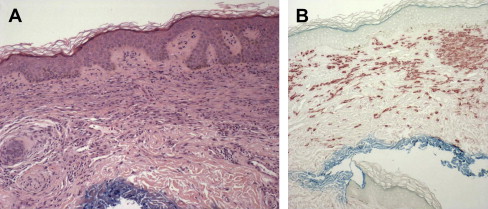
CM
Cutaneous MC infiltration can be recognized on hematoxylin and eosin staining ( Fig. 4 A); however, MCs are better visualized using basic dyes, such as Giemsa, toluidine blue, and Astra blue, or by immunocytochemical stains that reveal tryptase and chymase or histochemical techniques with chloroacetate esterase (see Fig. 4 B). In CM, MCs are spindle shaped or spherical and stain positively for both chymase and tryptase. There are 4 patterns of dermal MC infiltrate that have been described: (1) perivascular in the papillary body and upper dermis; (2) sheetlike within the papillary body and upper reticular dermis; (3) interstitial; and (4) nodular. There is a partial correlation between these 4 histologic patterns and the clinical presentations of CM. However, clinical features are related more to the density of the MC infiltrate. For example, the nodular pattern is found in nodular CM but can also be present in larger lesions associated with UP. The pattern of involvement does not predict the course of the disease or risk of systemic involvement. In CM, the number of MCs in the papillary dermis can be 10-fold higher than for the number found in normal skin ; however, the number required to diagnose CM has not been standardized. This point can make it difficult to differentiate between CM with low numbers of MCs and inflammatory conditions associated with an MC infiltrate, such as urticaria or atopic dermatitis. However, the presence of lymphocytes and other inflammatory cells is helpful in distinguishing non-CM inflammatory conditions from CM.