Ninety percent or more of kidney cancers are believed to be of epithelial cell origin, and are referred to as renal cell carcinoma (RCC), which are further subdivided based on histology into clear-cell RCC (75%), papillary RCC (15%), chromophobe tumor (5%), and oncocytoma (5%). Some genes confer an increased risk of these various histologic RCC subtypes. In practice, there is some overlap among the histologic subtypes, and there are some shared molecular features among these tumor types. This review focuses primarily on the most common form of RCC, clear-cell renal carcinoma, noting some recent advances in the other histologic subtypes.
Kidney cancer is one of the 10 most common forms of cancer in both men and women. Ninety percent or more of these cancers are believed to be of epithelial cell origin, and are referred to as renal cell carcinoma (RCC). RCCs can be further subdivided, based on their histologic appearance, into clear-cell renal carcinomas (∼75%), papillary renal carcinomas (15%), chromophobe tumors (5%), and oncocytomas (5%). Studies of hereditary kidney cancer families led to the identification of genes that, when mutated in the germline, confer an increased risk of these various histologic RCC subtypes and hence a glimpse at the molecular circuits that are deregulated in these different forms of RCC. In practice, there is some overlap among the histologic subtypes (eg, a tumor with predominantly clear-cell features might contain areas more typical of papillary RCC). Similarly, there are some shared molecular features among these tumor types (see later discussion). This review focuses primarily on the most common form of RCC, clear-cell renal carcinoma, while making note of some recent advances in the other histologic subtypes.
Clear-cell RCC genetics: role of the VHL tumor suppressor gene
von Hippel-Lindau (VHL) disease is a hereditary cancer syndrome characterized by an increased risk of clear-cell RCC, retinal and central nervous system hemangioblastomas, and pheochromocytomas. Individuals with this disorder carry a defective copy of the VHL tumor suppressor gene, typically inherited from either parent but occasionally resulting from a de novo mutation. Tumor development in this setting is linked to inactivation of the remaining wild-type allele in a susceptible cell, either as a result of deletion, intragenic mutation, or hypermethylation.
As might be predicted from this knowledge, VHL inactivation is also very common in sporadic clear-cell RCC. In most series about 50% of sporadic clear-cell RCC are unable to produce the normal (wild-type) VHL tumor suppressor protein (pVHL). Typically this reflects a sporadic intragenic mutation of one copy of the VHL gene, which is located on chromosome 3p, in conjunction with loss of the remaining copy because of a large deletion affecting the other 3p arm. In other tumors this loss of wild-type pVHL is the consequence of transcriptional silencing of both the maternal and paternal copies of the VHL gene as a result of hypermethylation. A third group of clear-cell RCC have a gene expression signature typical of VHL inactivation without demonstrable VHL mutations or hypermethylation. These tumors might theoretically have nonallelic mutations that indirectly affect VHL function, although to date no such mutations have been identified.
In light of these considerations one would expect that the frequency of chromosome 3p loss would approximate the frequency of intragenic VHL mutations in clear-cell RCC; however, this is not the case. In particular, the frequency of chromosome 3p is consistently higher (often >90%) than the frequency of intragenic VHL mutations. There are several, nonmutually exclusive, possibilities to explain this conundrum. First, the failure to detect intragenic mutations in some tumors might be due to technical factors (ie, reflecting “false negatives”). If true, this would provide an alternative explanation for the existence of “ VHL +/+” clear-cell RCC that display a VHL −/− gene expression signature. Indeed, there is some evidence that new sequencing technologies will reveal a higher VHL mutation rate in clear-cell RCC. Second, chromosome 3p might harbor additional clear-cell RCC suppressor genes. In this regard, frequent inactivating mutations of the PBRM1 gene, located on chromosome 3p21, were recently reported in clear-cell RCC. This gene encodes a protein involved in the control of DNA packing into chromatin. Finally, it is possible that loss of one copy of VHL (haploinsufficiency) promotes clear-cell RCC in some settings, although there are presently no laboratory data to support the idea that VHL haploinsufficiency measurably alters the molecular pathways described later in this article.
Although VHL inactivation is an important event in the pathogenesis of many clear-cell RCC, it is not sufficient to cause this disease. This aspect has been most clearly documented by examining the kidneys of VHL patients, which often contain hundreds of preneoplastic lesions that have sustained loss of the remaining wild-type VHL allele, and yet few carcinomas. Similarly, VHL inactivation in the murine kidney is not sufficient to cause RCC. It appears that additional genetic events, occurring stochastically, are required to convert a VHL −/− preneoplastic lesion into clear-cell RCC.
Indeed, several nonrandom chromosomal changes, in addition to chromosome 3p loss, have been described in both sporadic and hereditary (VHL disease-associated) clear-cell RCC. These alterations include frequent loss of chromosome 14q and gain of chromosome 5q. It is interesting that some clear-cell RCC contain unbalanced translocations of chromosomes 3 and 5, leading to loss of 3p and gain of 5q. Recurrent amplifications and deletions in clear-cell RCC presumably harbor oncogenes and tumor suppressor genes, respectively, which play roles in the pathogenesis of clear-cell RCC.
Direct sequencing of kidney cancer genomes provides information that is complementary to technologies that measure changes in copy number (ie, amplifications and deletions). This approach has revealed that several chromatin-modifying enzymes, including the SETD2 histone methyltransferase, the JARID1C (also known as KDM5C) histone demethylase, and the UTX (also known as KMD6A) histone demethylase, are sometimes mutated in clear-cell RCC.
Functions of the von Hippel-Lindau Tumor Suppressor Protein
The VHL tumor suppressor gene encodes 2 proteins by virtue of 2 alternative, in-frame, start codons. The shorter form lacks 53 N-terminal amino acid residues that are present in the long form. For simplicity, both forms here are referred to as “pVHL” because they behave similarly in most biochemical and biological assays performed to date, and because all of the VHL mutations identified in clear-cell carcinoma so far would be predicted to affect both pVHL isoforms. pVHL resides primarily in the cytoplasm but dynamically shuttles between the nucleus and the cytoplasm, and has also been detected in association with the endoplasmic reticulum and mitochondria.
pVHL is the substrate recognition component of an E3 ubiquitin ligase complex that regulates hypoxia-inducible factor (HIF) ( Fig. 1 ). HIF is a heterodimeric DNA-binding transcription factor consisting of an unstable α subunit and a stable β subunit. When oxygen is present the α subunit becomes hydroxylated on one (or both) of two conserved prolyl residues by members of the EglN (also called PHD) prolyl hydroxylase family. Once prolyl hydroxylated, the α subunit is recognized by pVHL, polyubiquitinated, and destroyed by the proteasome. Under low oxygen conditions, or in cells lacking wild-type pVHL, the α subunit is stabilized, dimerizes with a β subunit, and transcriptionally activates 100 to 200 genes, many of which are involved in acute or chronic adaptation to hypoxia. Examples of such genes include VEGF , which promotes angiogenesis, EPO , which promotes erythropoiesis, and a suite of genes that promote glucose uptake and glycolysis.

There are 3 HIFα genes in the human genome (HIF1α, HIF2α, and HIF3α). HIF1α is ubiquitously expressed whereas the expression of HIF2α is more restricted. Both HIF1α and HIF2α can bind to specific DNA sequences (hypoxia-responsive elements) and activate transcription. HIF1α and HIF2α share many target genes, but it is also becoming increasingly clear that some genes are preferentially activated by one or the other. Multiple HIF3α splice variants have been described, many of which might actually serve to interfere with the function of HIF1α and HIF2α.
Many lines of evidence point to a pivotal role of HIF, and especially HIF2α, in pVHL-defective clear-cell RCC. The risk of developing clear-cell RCC linked to germline VHL mutations mirrors the degree to which those mutations deregulate HIF. In preclinical models HIF2α, but not HIF1α, can override the tumor suppressor activity of pVHL and elimination of HIF2α, but not HIF1α, is sufficient to suppress tumor formation by pVHL-defective clear-cell renal carcinoma cells. Moreover, HIF2α appears to account for much or all of the pathology observed in mice that have been engineered to lack pVHL in certain tissues. In further support of the importance of HIF2α, pVHL-defective clear-cell RCC produce both HIF1α and HIF2α or, informatively, HIF2α alone. Close examination of preneoplastic renal lesions in patients with VHL disease suggests that the appearance of HIF2α, rather than HIF1α, heralds malignant transformation, and a recent study linked the risk of developing clear-cell RCC to single-nucleotide polymorphisms affecting HIF2α. Collectively these studies underscore that pVHL-defective clear-cell RCC are driven, at least partly, by a surfeit of HIF2α activity.
The EglN prolyl hydroxylases require, in addition to oxygen, the metabolite 2-oxoglutarate (also called α-ketoglutarate) to hydroxylate HIFα. During the hydroxylation reaction, 2-oxoglutarate is converted to succinate. Succinate can be converted by succinate dehydrogenase (SDH) to fumarate, which in turn can be converted to malate by fumarate hydratase (FH). Inactivating SDH and FH mutations, which lead to an increase in succinate and fumarate, respectively, have been linked to an increased risk of RCC. This link has been most clearly demonstrated by the increased risk of papillary RCC observed in kindreds bearing FH mutations. Succinate and fumarate can inhibit EglN activity in vitro and in vivo, leading to HIF activation. These findings suggest that HIF deregulation, whether due to inactivation of VHL, SDH, or FH, is a feature of RCC.
pVHL has several other functions that, although incompletely understood at a biochemical level, seem to be at least partly HIF-independent and which might be relevant to tumorigenesis ( Fig. 2 ). These functions include roles in the assembly of an extracellular fibronectin matrix, receptor internalization, microtubule stability, nerve growth factor signaling, senescence, and maintenance of the primary cilium, which is a specialized structure on the surface of cells that serves as a sensory antenna (eg, responding to changes in adjacent fluid dynamics). This latter function is particularly intriguing because visceral cysts, including renal cysts, are a prominent feature of VHL disease and are also seen in several other unrelated diseases, such as Bardet-Biedl syndrome, that affect the primary cilium (so-called ciliopathies). Whether these disparate pVHL functions relate to its function as part of an E3 ubiquitin ligase is not known. In this regard it is perhaps noteworthy that the other core components of the pVHL ubiquitin ligase complex, such as elongin B, elongin C, and Cul2, are not frequently mutated in clear-cell RCC. This fact might reflect the importance of pVHL-independent functions of these proteins in cellular homeostasis or the importance of ubiquitin ligase–independent functions of pVHL in tumorigenesis (or both).
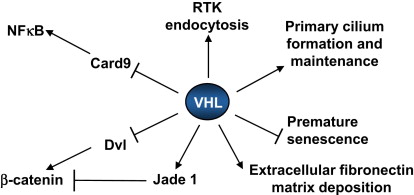
Recent studies suggest that pVHL regulates, at least indirectly, the nuclear factor κB (NF-κB) transcription factor. pVHL acts as an adaptor molecule that promotes the inhibitory phosphorylation of the NF-κB agonist Card9 by casein kinase II, thereby suppressing NF-κB activity. In addition, HIF itself can activate NF-κB in some contexts whereas NF-κB can promote the transcription of HIF1α. In keeping with the aforementioned, NF-κB activity is increased in clear-cell RCC and might contribute to the resistance of this tumor to many forms of therapy. In addition, HIF and NF-κB share several target genes and hence might cooperate to promote the development of clear-cell RCC.
pVHL, in addition to regulating HIF and NF-κB, regulates the Wnt signaling pathway and hence the activity of the oncogenic transcription factor β-catenin. Several nonmutually exclusive mechanisms to account for suppression of Wnt signaling by pVHL have been proposed. pVHL promotes the ubiquitination of the Wnt signaling protein Disheveled (Dvl), which then interacts with SQSTM1/p62 and is subsequently degraded by the autophagy-lysosome pathway. pVHL also stabilizes the candidate tumor suppressor protein Jade-1, which in turn is capable of targeting β-catenin for proteasomal degradation. Finally, HIF1α itself modulates Wnt/β-catenin signaling in hypoxic embryonic stem (ES) cells by enhancing β-catenin activation and expression of the downstream effectors LEF-1 and TCF-1. Thus pVHL inhibits, directly or indirectly, the oncogenic transcription factors HIF2α, NF-κB, and β-catenin. Conversely, Wnt signaling can affect VHL expression in some tissues.
HIF as a therapeutic target in RCC
The central role of HIFα, and particularly HIF2α, in pVHL-defective clear-cell RCC has spurred interest in the development of HIF antagonists for this disease. Unfortunately, however, there are very few examples of drug-like small organic molecules that are capable of inhibiting the function of DNA-binding transcription factors, with the notable exception of the steroid hormone receptors.
Inhibition of the mammalian target of rapamycin (mTOR) kinase decreases the transcription and translation of HIF1α, thereby lowering HIF1α protein levels. This process might account for the observation that two rapamycin-like mTOR inhibitors (“rapalogs”), temsirolimus and everolimus, have activity in the treatment of clear-cell RCC ( Fig. 3 ). Unfortunately, these two drugs primarily inhibit mTOR when it is in the multiprotein complex called TORC1 rather than in the TORC2 complex. This aspect is important because HIF2α, which as noted is the HIFα paralog most closely linked to the pathogenesis of pVHL-defective clear-cell RCC, appears to be more sensitive to TORC2 loss than to TORC1 loss. Preclinical studies support the idea that adenosine triphosphate–competitive mTOR inhibitors, which inhibit both TORC1 and TORC2, will more effectively downregulate HIF2α and suppress tumor growth than do rapalogs.
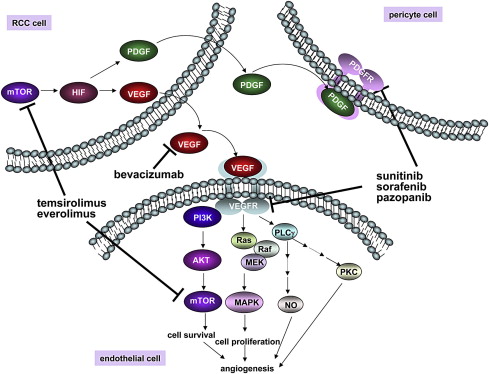
Several other drugs have been identified that, at least indirectly, are capable of downregulating HIF activity in cells. A cautionary note, however, is that HIFα, by virtue of having a high metabolic turnover rate (even in pVHL-defective cells), is very sensitive to decreases in its rate of transcription and translation. HIFα will therefore disappear more rapidly in response to drugs that are generally toxic than do many housekeeping proteins that are commonly used as specificity controls in evaluating putative HIF inhibitors. In short, it is possible that some drugs that have been reported to inhibit HIF actually have an inhibitory effect on transcription or translation in general, and would affect a variety of short-lived proteins.
HIF as a therapeutic target in RCC
The central role of HIFα, and particularly HIF2α, in pVHL-defective clear-cell RCC has spurred interest in the development of HIF antagonists for this disease. Unfortunately, however, there are very few examples of drug-like small organic molecules that are capable of inhibiting the function of DNA-binding transcription factors, with the notable exception of the steroid hormone receptors.
Inhibition of the mammalian target of rapamycin (mTOR) kinase decreases the transcription and translation of HIF1α, thereby lowering HIF1α protein levels. This process might account for the observation that two rapamycin-like mTOR inhibitors (“rapalogs”), temsirolimus and everolimus, have activity in the treatment of clear-cell RCC ( Fig. 3 ). Unfortunately, these two drugs primarily inhibit mTOR when it is in the multiprotein complex called TORC1 rather than in the TORC2 complex. This aspect is important because HIF2α, which as noted is the HIFα paralog most closely linked to the pathogenesis of pVHL-defective clear-cell RCC, appears to be more sensitive to TORC2 loss than to TORC1 loss. Preclinical studies support the idea that adenosine triphosphate–competitive mTOR inhibitors, which inhibit both TORC1 and TORC2, will more effectively downregulate HIF2α and suppress tumor growth than do rapalogs.
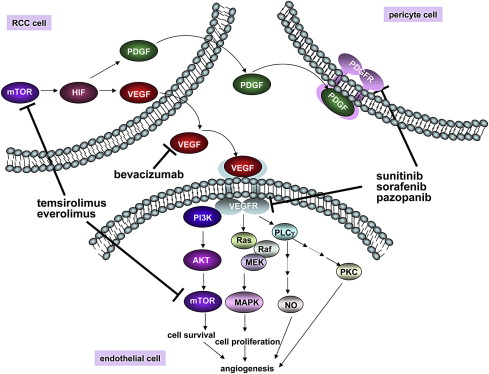
Several other drugs have been identified that, at least indirectly, are capable of downregulating HIF activity in cells. A cautionary note, however, is that HIFα, by virtue of having a high metabolic turnover rate (even in pVHL-defective cells), is very sensitive to decreases in its rate of transcription and translation. HIFα will therefore disappear more rapidly in response to drugs that are generally toxic than do many housekeeping proteins that are commonly used as specificity controls in evaluating putative HIF inhibitors. In short, it is possible that some drugs that have been reported to inhibit HIF actually have an inhibitory effect on transcription or translation in general, and would affect a variety of short-lived proteins.
HIF-responsive gene products
Vascular Endothelial Growth Factor
Kidney cancers are highly angiogenic tumors. In fact, renal angiograms were often used to diagnose this neoplasm prior to the availability of computed tomography. The robust induction of angiogenesis by these tumors appears to be due, at least partly, to overproduction of vascular endothelial growth factor (VEGF), which is the product of a HIF-responsive gene. Accordingly, VEGF is normally induced by hypoxia but is constitutively produced in pVHL-defective tumors. There has been a suggestion that VEGF might also act in an autocrine fashion to directly stimulate pVHL-defective tumors cells, although this idea remains controversial.
Many VEGF inhibitors, including the neutralizing antibody bevacizumab and small molecules directed against the VEGF receptor VEGFR2 (also called KDR), such as sunitinib, sorafenib, and pazopanib, have been approved for the treatment of kidney cancer based on randomized clinical trial data (see Fig. 3 ). In most studies about 70% of patients with kidney cancer will achieve some degree of tumor shrinkage when treated with a VEGF inhibitor, although the percentage of people achieving a partial response, as measured by RECIST criteria, varies considerably among the different agents. This finding might reflect differences in potency as well as the contribution of fortuitous “off-target” effects (see also later discussion). VEGF inhibitors have been approved for the treatment of metastatic kidney cancer based primarily on a significant improvement in terms of progression-free survival and a trend toward improved survival. The actual benefits of these agents in terms of overall survival are difficult to determine because control patients have typically been allowed to cross over to VEGF inhibitors at the time of disease progression.
There are conflicting reports as to whether VHL gene status influences the response to the existing VEGF inhibitors (ie, whether it can be used as a predictive biomarker). These disparities might be due, at least partly, to technical factors such as the different methodologies used to identify VHL mutations in the different studies. In addition, the predictive value of VHL gene status might be diluted by the existence of VHL +/+ tumors that are phenotypically pVHL-defective, as described earlier. Suffice it to say that VHL gene status does not appear to be a robust predictive biomarker at present and should therefore not be used to guide therapeutic decision making, especially given the paucity of alternative agents.
Several second-generation KDR inhibitors that display improved potency and specificity are currently in development. It is hoped that these attributes will lead to enhanced clinical efficacy, reduced toxicity, and a greater ability to be combined with other agents. With respect to activity, however, there is already some evidence that there might be a limit to how completely VEGF signaling can be safely blocked in man. Microangiopathic changes, presumably reflecting damage to normal endothelial cells, have been reported in patients treated with a combination of two VEGF inhibitors.
The vast majority of patients treated with a VEGF inhibitor will eventually become resistant to that agent and experience disease progression. Of interest, some of these patients will respond to a second, unrelated, VEGF inhibitor, suggesting that the patient’s tumor remained VEGF dependent. In other cases resistance to VEGF inhibition might, based on preclinical models, reflect activation of alternative angiogenic factors, such as fibroblast growth factor and interleukin (IL)-8, or to the ability of tumor cells to undergo a reversible epithelial-mesenchymal transition.
Platelet-Derived Growth Factor
Platelet-derived growth factor (PDGF) B is another HIF target gene linked to angiogenesis. PDGF, which activates the PDGF receptor (PDGFR), stimulates the coverage of newly sprouting blood vessels by pericytes. In preclinical models, proliferating endothelial cells are less susceptible to VEGF withdrawal once they are provided with survival signals by such pericytes. Accordingly dual inhibition of VEGF and PDGF should, in principle, be more effective than VEGF blockade alone. Several of the currently available KDR inhibitors do, in fact, have considerable activity against PDGFR. Whether this indeed contributes to their clinical activity is not known, however.
Transforming Growth Factor α and Epidermal Growth Factor Receptor
HIF induces the expression of the mitogenic transforming growth factor α (TGFα) and, at least indirectly, upregulates and activates its receptor, epidermal growth factor receptor (EGFR). In addition, pVHL loss may potentiate EGFR signaling by decreasing its rate of internalization and recycling at the cell membrane. EGFR is overexpressed and active in kidney cancers, and mouse models support an important role for EGFR in pVHL-defective tumor growth. Indeed, silencing EGFR suppresses tumor formation by VHL −/− RCC cells. Nonetheless, the activity of EGFR inhibitors in the treatment of human kidney cancer has thus far been very disappointing. A trivial explanation for this lack of a clinical response would be a failure to achieve a sufficient pharmacodynamic effect with the EGFR inhibitors tested so far. Another possibility relates to the potential importance of collateral signaling pathways. In particular, activation of c-Met confers partial resistance to EGFR inhibitors and, as discussed later, is frequently activated in pVHL-defective tumors. Murine hepatocyte growth factor (HGF), the ligand for c-Met, does not activate human c-Met. For this reason, the murine tumor xenograft studies performed to date with kidney cancer lines might have underestimated the potential influence of c-Met on EGFR dependence. A third, nonmutually exclusive, possibility is that the available kidney cancer lines became dependent on EGFR during adaptation to cell culture conditions.
Cyclin D
In renal epithelial cells, but not in the other epithelial cells tested thus far, HIF upregulates the expression of the cyclin D1 oncoprotein. Cyclin D1 promotes cell cycle progression by activating Cdk4 and Cdk6, which phosphorylate and thereby inactivate the pRB tumor suppressor protein. Cdk6 is one of many genes located on a large chromosome 7 kidney cancer amplicon. It is interesting that pVHL-defective clear-cell RCC appear to be hypersensitive to Cdk6 inhibition compared with isogenic, pVHL-proficient, control cells. Several Cdk4/6 inhibitors are in clinical development.
Hepatocyte Growth Factor and c-Met
Several reports indicate that signaling by c-Met is enhanced by pVHL loss, although the exact mechanisms responsible are in dispute. Some studies have reported that c-Met protein levels are increased by hypoxia and HIF, whereas other studies have focused on the effect of pVHL loss on signaling events downstream of c-Met. Of interest is that c-Met is mutationally activated in a subset of familial papillary renal cancers and can, at least in certain settings, activate HIF. This finding suggests that there is potential cross-talk between HIF and c-Met, and that there are molecular similarities between clear-cell and papillary renal cancer. Both c-Met and its ligand HGF are located on the large chromosome 7 amplicon described earlier, and pVHL-defective cells appear to be hypersensitive to c-Met loss. c-Met also plays a role in angiogenesis in addition to its potential cell autonomous role in clear-cell RCC. All of these considerations support the testing of c-Met inhibitors in clear-cell RCC.
Insulin-Like Growth Factor 2 and Insulin-Like Growth Factor 1 Receptor
HIF regulates the expression of insulin-like growth factor 2 (IGF2). In addition, pVHL suppresses, in a HIF-independent manner, insulin-like growth factor 1 receptor (IGF1R) protein levels and downstream signaling. The former has been linked to suppression of SP1 and sequestration of the HuR RNA-binding protein, and the latter to inhibition of protein kinase Cδ. IGF1R signaling can, in turn, upregulate HIF (particularly HIF1α). Downregulation of IGFR sensitizes clear-cell RCC to conventional cytotoxics and to rapamycin-like drugs. With respect to the latter, treatment of many cell types with rapalogs leads to a paradoxic increase in Akt activity due to loss of feedback inhibition of receptor tyrosine kinases such as IGFR, providing a rationale for combining rapalogs with IGFR inhibitors.
Stromal Cell–Derived Factor and C-X-C Chemokine Receptor Type 4
Hypoxia and HIF increase the expression of the cytokine stromal cell–derived factor (SDF) and its receptor C-X-C chemokine receptor type 4 (CXCR4). This cytokine-receptor pair has been implicated in tumor growth and invasion in other settings as well as in vasculogenesis, the process by which de novo blood vessels are formed through the recruitment of bone marrow–derived cells such as endothelial cell precursors and certain hematopoietic cells. Preclinical studies in other tumor types suggest that blockade of VEGF and SDF would have additive effects.
Interleukin-6
IL-6 functions as an autocrine growth factor in kidney cancer, and its expression is suppressed by pVHL. Binding of IL-6 to its receptor, IL-6R, triggers receptor dimerization and phosphorylation by members of the Janus kinase families. Activation of IL-6R leads to STAT3 activation that promotes RCC cell proliferation. Primary clear-cell RCCs produce high levels of IL-6 and frequently express IL-6R. In addition, expression of IL-6R correlates with high serum IL-6 concentration and metastatic progression in this setting. A clinical trial conducted in patients with progressive metastatic kidney cancer showed that CNOT328, a chimeric murine-human monoclonal antibody against IL-6, stabilized disease in more than 50% of the cases.
RTK-Like Orphan Receptor 2
RTK-like orphan receptor 2 (ROR2) was identified in a biochemical screen for kinases that are induced and activated by pVHL loss in renal carcinoma cells. ROR2 has an intracellular tyrosine kinase domain and an extracellular frizzled-like cysteine-rich domain that can act as a receptor for Wnt ligands. In preclinical models ROR2 promotes tumor cell invasiveness, at least partly through the upregulation of matrix metalloproteinase 2, soft agar growth, and orthotopic tumor formation. These studies raise the possibility that targeting Wnt ligands or ROR2 itself will favorably alter the natural history of pVHL-defective clear-cell RCC.
Aurora Kinase A
pVHL inactivation leads to HIF-dependent induction of Nedd9 (also called renal carcinoma antigen NY-REN-1) and Aurora Kinase A, which colocalize at the centrosome. Aurora A activation is Nedd9 dependent. Aurora A destabilizes ciliary microtubules through its ability to phosphorylate, and thereby activate, HDAC6, which deacetylates tubulin. Suppression of Aurora Kinase A partially restores primary cilium formation, and impairs motility, in pVHL defective clear-cell RCC.
Angiopoietin 4
The angiopoietins (Ang) and their receptor, TIE2, play an important role in angiogenesis. Ang1 and Ang4 act as TIE2 agonists, and Ang2 is a TIE2 antagonist. Activation of TIE2 promotes the stabilization and maturation of vessels, whereas inhibition of TIE2 destabilizes blood vessels. In the latter context VEGF promotes sprouting of new blood vessels from the destabilized vessel wall, whereas loss of VEGF promotes endothelial cell death. These biological complexities have made it difficult to predict, a priori, the net effect of manipulating this system with respect to tumor angiogenesis. Nonetheless, pVHL has been reported to decrease the expression of Ang4 under normoxia, and blocking TIE2 blunts endothelial tube formation in response to pVHL-defective conditioned media in vitro. The relationship between pVHL and Ang2 appears to be more complex, with pVHL seemingly required for the downregulation of Ang2 by hypoxia. These findings support the exploration of TIE2 antagonists in conjunction with VEGF inhibitors for the treatment of clear-cell RCC.
Lactate Dehydrogenase A and Carbonic Anhydrase
HIF stimulates glycolysis and inhibits oxidative phosphorylation. The shift to glycolysis is accompanied by HIF-dependent increases in lactate dehydrogenase A, which converts pyruvate to lactate and carbonic anhydrases IX and XII, which help cells to accommodate the increased lactic acid load. These enzymes can be inhibited with drug-like small molecules, and preclinical studies suggest that such compounds would preferentially kill pVHL-defective tumor cells.
Histone Methylases and Demethylases
HIF induces the expression of several histone demethylases including JMJD1A, JMJD2B, and JARID1B. JMJD1A is, as expected, overexpressed in RCC, and its downregulation modestly suppresses tumor growth in vivo in preclinical models. JARID1B (also called PLU-1) has been implicated as an oncoprotein in breast cancer, raising the possibility that it promotes RCC growth as well. The identification of mutations affecting histone methylation in RCC also suggests that one or more enzymes that add or remove specific histone methyl marks will emerge as potential drug targets for this disease.
Related posts:
 Renal Cell Carcinoma
Renal Cell Carcinoma
 mTOR Inhibitors in Advanced Renal Cell Carcinoma
Future Directions in Renal Cell Carcinoma: 2011 and Beyond
mTOR Inhibitors in Advanced Renal Cell Carcinoma
Future Directions in Renal Cell Carcinoma: 2011 and Beyond
 Management of Treatment-Related Toxicity with Targeted Therapies for Renal Cell Carcinoma: Evidence-Based Practice and Best Pr actices
Small Renal Masses: Risk Prediction and Contemporary Management
mTOR Inhibitors in Advanced Renal Cell Carcinoma
Management of Treatment-Related Toxicity with Targeted Therapies for Renal Cell Carcinoma: Evidence-Based Practice and Best Pr actices
Small Renal Masses: Risk Prediction and Contemporary Management
mTOR Inhibitors in Advanced Renal Cell Carcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


