Clinical characteristic
Diagnosis
Major features
Facial angiofibromas or forehead plaque
Definite TSC:
Either 2 major features or 1 major features plus 2 minor features
Probable TSC:
One major plus 1 minor feature
Nontraumatic ungual or periungual fibroma
Hypomelanotic macules (≥3)
Shagreen patch migration lines
Multiple retinal nodular hamartomas
Cortical tubera
SENs
SEGAs
Cardiac rhabdomyoma, single or multiple
LAM, renal angiomyolipoma, or bothb
Minor features
Multiple, randomly distributed pits in dental enamel
Possible TSC:
Either 1 major feature or ≥2 minor features
Hamartomatous rectal polypsc
Bone cysts
Cerebral white matter radial migration linesa,d
Gingival fibromas
Nonrenal hamartomab
Retinal achromic patch
“Confetti”-like skin lesions
Multiple renal cystsb
6.2.2 Epidemiology and Molecular Genetics of TSC
TSC is a rare, autosomal-dominant, single-gene disorder with an incidence of 1 in 6800 to 1 in 17,300 live births, resulting in a total of one million individuals estimated to be affected globally [14, 15]. It is genetically transmitted via mutations in TSC1 or TSC2, although a somatic second-“hit” mutation in the unaffected TSC allele, in addition to the germline mutation, is required for tumor development [3]. Thus, consistent with their role as tumor suppressor genes [16], inactivation of both copies of TSC1 or TSC2 leads to the formation of hamartomas [2, 3]. In about 85 % of all patients with TSC, mutation in either TSC1 or TSC2 can be identified [17]. Nearly 70–80 % of TSC cases are sporadic [16], whereas one-third of cases [18] are a familial mutation, identified in either TSC1 or TSC2, which the patients have inherited from their parents [2, 3]. However, 15–20 % of patients with a clinical diagnosis of TSC have no mutation identified (NMI) in TSC1 or TSC2 [2, 6]. Lack of sensitivity in mutation detection methods, incomplete assessment of other regions such as introns and other regulatory sequences within the TSC1/TSC2 locus, and tissue mosaicism, in which TSC mutations are not universally expressed in all cells in the affected individual, are some of the reasons that may account for patients with NMI [6].
It has been difficult to correlate specific TSC disease phenotypes with the genotype due to the fact that the same germline mutation can result in phenotype variability as a result of the type and time of the second-hit mutation in the unaffected allele [16]. TSC2 mutations are three times more frequent than TSC1 mutations in patients with TSC and are associated with a more severe phenotype [2, 17]. Despite the differences in phenotype associated with TSC1 and TSC2 mutation carriers, clinical prognosis is not possible based on the type of TSC2 mutation, as the symptoms vary greatly even among patients with identical TSC2 mutations [19]. The only exception is in the case of patients with contiguous deletions in TSC2 and PKD1, also located on chromosome 16p13.3, who will develop severe polycystic kidney disease (PKD) early on in their lives [3, 10].
6.2.3 Central Role of mTOR Signaling in TSC
Dysregulation of mTOR activity due to TSC mutations results in the persistent phosphorylation of its downstream effectors such as p70 ribosomal S6 kinase 1 (S6K1, an activator of translation) and eukaryotic initiation factor 4E-binding protein 1(4E-BP1, an inhibitor of translation initiation) [1]. Indeed, in fibroblasts derived from TSC1 null embryos, mTOR signaling is constitutively active, as seen by the elevated phosphorylation of S6K1 [13]. In addition, reduction of TSC2 protein levels by TSC2 RNA interference (RNAi) has been shown to increase the phosphorylation of S6K1, which can be abolished by expression of wild-type TSC2 DNA, but not by TSC2 DNA-carrying mutations that are commonly seen in patients with TSC [13]. Moreover, TSC2-null cells and cells in which TSC2 has been downregulated by RNAi showed increased levels of HIF-1α, which in turn resulted in upregulation of transcription of vascular endothelial growth factor (VEGF), a key player in angiogenesis [20]. Importantly, the increase in HIF-1α in TSC2-null cells could be reversed by either inhibition of the mTOR pathway through rapamycin treatment or by reconstitution of TSC2-null cells with wild-type TSC2, but not by TSC2 mutants carrying disease-associated mutations [20]. Similarly, rapamycin treatment was able to normalize the increased production of VEGF secretion seen in TSC1– and TSC2-null fibroblasts [21]. These data provide biochemical evidence linking TSC1/TSC2 to dysregulated mTOR signaling as seen by constitutively activated S6K1 and its relationship to TSC mutations in individuals with TSC.
6.3 Clinical Manifestations of TSC and Dysregulated mTOR Signaling
6.3.1 Neurological Manifestations
Individuals affected with TSC exhibit a broad range of central nervous system (CNS) manifestations [3]. The structural abnormalities in the brain include lesions such as cortical tubers, subependymal nodules (SENs), and subependymal giant cell astrocytomas (SEGAs) [3]. CNS lesions typically seen in TSC vary over the lifetime of the patient, with cortical tubers often detectable prenatally (i.e., at 20–26 weeks of gestation), SENs identifiable in childhood, and SEGAs developing in childhood and beyond [2, 3]. Cortical tubers occur in over 80 % of patients with TSC and are characterized by loss of the typical six-layered structure of the cerebral cortex [15]. SENs occur in 88–95 % of patients with TSC and develop near the wall of the cerebral ventricles [22]. They usually remain dormant throughout life but have the potential to increase in size and develop into SEGAs [15]. SEGAs occur in approximately one of every ten individuals with TSC [22]. Growth of these tumors can obstruct cerebrospinal fluid flow, leading to hydrocephalus, increased intracranial pressure, and, potentially, death [2].
Dysregulated mTOR activity, as evidenced by enhanced phosphorylation of S6, is seen in the enlarged neurons of the human tubers [23, 24]. In a mouse model, controlled loss of heterozygosity in TSC1 resulted in focal brain malformations that closely modeled human tubers [18]. These tuber-like lesions displayed enhanced mTOR signaling as evidenced by the elevated levels of phospho-S6. Aberrant mTOR signaling is also evident in SEGA cells with biallelic mutations in TSC1/TSC2, showing high levels of phospho-S6K, phospho-S6, and phospho-Stat3 (all proteins), downstream of mTOR activation [25]. In yet another mouse model, conditional deletion of TSC1 allele in newborns resulted in the formation of nodules along the subependymal zone and SEGA-like lesions [18]. mTOR hyperactivity in these TSC1-deleted neurons was evidenced by an increase in phospho-S6 staining and an increase in cell size, compared with TSC1-positive neurons [26].
Patients with TSC also display a wide spectrum of neurological symptoms [27]. Approximately 85 % of children and adolescents with TSC have CNS manifestations such as infantile spasms, epilepsy, cognitive impairment, and autism [3]. Challenging behavioral problems such as attention-deficit/hyperactivity disorder (ADHD), aggression, rage, hyperactivity, obsessive/repetitive behavior, and intellectual disability are also seen [27, 28].
mTOR signaling in the CNS is important for synaptogenesis, axon myelination and growth, dendrite morphogenesis, neurotransmitter-receptor expression, and neuronal growth [29]. Loss of a single copy of TSC1/TSC2 resulted in neurocognitive defects, emphasizing the role of TSC1/TSC2 and the mTOR pathway in the maintenance and regulation of connectivity and communication in the CNS, which is mediated by structural components such as dendrites, synapses, and axons [18]. mTOR signaling has been shown to play a role in the dendritic morphogenesis in vitro in several studies, and inhibition of the mTOR pathway using rapamycin or RNAi knockdown of mTOR or S6K eliminated these morphogenetic effects [18]. Overexpression of TSC1 or TSC2 in cultured neurons suppressed the mTOR activity and axon formation, whereas depletion of TSC1 or TSC2 via RNAi promoted growth of multiple axons [18]. Thus, dysregulated mTOR signaling that causes changes in cell growth, proliferation, neurotransmitter-receptor and ion-channel expression, neuronal structure, and synaptic plasticity could potentially lead to TSC-associated epileptogenesis, autism, and cognitive impairment [29].
6.3.2 Renal Lesions
Renal manifestations are the third most common clinical feature in patients with TSC. Four kinds of renal lesions can occur: angiomyolipomas, isolated renal cysts, ADPKD, and RCC. Angiomyolipomas and isolated renal cysts are the two most common renal lesions observed in patients with TSC [30].
Angiomyolipomas are vascular tumors composed of dysplastic or abnormal blood vessels (angio), smooth-muscle cells (myo), and fat (lipoma), and they can range from being asymptomatic to causing renal failure [30, 31]. The incidence of angiomyolipomas in patients with TSC is estimated from several studies to range from 48 to 80 %, depending on the age population being studied [6, 32]. Several scientific reports have demonstrated dysregulated mTOR signaling in patient-derived angiomyolipomas. Loss of tuberin expression in angiomyolipoma samples correlated with increased phosphorylation of S6K1 and the ribosomal S6 protein in smooth-muscle cells in four out of five angiomyolipomas [33]. Similarly, high phospho-S6 was seen in both smooth-muscle and fat cells from angiomyolipomas in which loss of heterozygosity in either TSC1 or TSC2 was detected [34].
Approximately 30 % of patients with TSC associated with either the TSC1 or TSC2 mutations show presence of renal cystic disease [16]. In preclinical models, deletions in TSC2 have been shown to dysregulate renal cell polarity and effectuate the development of cysts [35]. In 2 % of all patients with TSC, large deletions in TSC2 also affect the PKD1 gene, which is adjacent to TSC2, resulting in an ADPKD phenotype with an early onset at birth or during childhood.
6.3.3 Pulmonary Manifestations
Lymphangioleiomyomatosis (LAM) is the primary pulmonary manifestation of TSC, occurring in 30–40 % of women with TSC [36]. LAM is characterized by the diffuse proliferation of abnormal smooth-muscle cells, micronodular pneumocyte hyperplasia, and cystic destruction of the lungs [37]. Pleural complications such as pneumothorax, chylous pleural effusions, and decreased pulmonary function measured as the forced expiratory volume in 1 s (FEV1) are some of the common clinical manifestations of LAM. LAM also occurs in women not affected with TSC, which is then referred to as sporadic LAM (S-LAM). While other manifestations of TSC are not associated with S-LAM, angiomyolipomas occur in about 60 % of women with S-LAM. In S-LAM, mutations are found in both alleles of TSC2 and are present in LAM cells and angiomyolipoma cells but not in normal lung or kidney cells [38]. Consistent with the lack of TSC1/TSC2, cells from LAM nodules from the lungs of patients have constitutively activated S6K1 and hyperphosphorylated ribosomal protein S6, indicative of a hyperactive mTOR pathway [36]. Primary cultures of smooth-muscle-α-positive LAM cells, derived from patients with S-LAM, also expressed high levels of phospho-S6, as well as its upstream kinase, S6K [36]. Reexpression of TSC2 in smooth-muscle-positive LAM cells inhibited the constitutively activated mTOR/S6K1 signaling. Together, these observations identify mTOR dysfunction due to loss of TSC1/TSC2 as one of the key mechanisms in the etiology and pathology of LAM.
6.3.4 Other Manifestations of TSC
In addition to CNS, renal, and pulmonary manifestations, patients with TSC may also develop cardiac rhabdomyomas (CR), which are benign tumors that may be focal or diffuse and infiltrating in character. These tumors develop within the cardiac cavities prenatally and regress spontaneously with age; however, these can sometimes be associated with cardiac arrhythmias [2]. Increased expression of mTOR, pS6K, and 4E-BP1 has been found in all CR samples and decreased TSC1/TSC2 expression in tumors versus normal heart tissues [39]. Additionally, in an animal model with tissue-specific knockdown of TSC1 in ventricular myocytes, mice developed cardiomyopathy and enlarged myocytes with increased phospho-S6 expression, similar to the human CR cells [40].
TSC is also associated with skin lesions such as hypomelanotic macules, shagreen patches, periungual or subungual fibromas, facial angiofibromas, and fibrous plaques, which can manifest on the skin, face, body, and nails of both children and adults [2]. Facial angiofibromas manifest in youth and early childhood and affect 70–80 % of patients with TSC [41]. Most skin lesions are asymptomatic, but they can be cosmetically bothersome and have a significant impact on a patient’s self-perception and quality of life [42]. Facial angiofibroma or white patches may affect the self-esteem of an individual and thus lead to withdrawal from social interaction [6].
6.4 mTOR Inhibition in TSC: Preclinical and Clinical Studies
Historically, treatment options for TSC have included lifelong monitoring and management of symptoms, primarily with invasive methods, rather than curing the underlying cause of the disease. The central role of mTOR signaling in the development of TSC makes it a rational therapeutic target for treatment of TSC. mTOR inhibitors, rapamycin (sirolimus/Rapamune) and everolimus (RAD001/Afinitor), have been extensively investigated in TSC-associated manifestations [7]. These agents are macrolide lactones that bind to intracellular immunophilin protein, FK-binding protein-12 (FKBP-12), to form an inhibitory complex, which in turn binds with high affinity to mTORC1, thereby inhibiting mTORC1. Everolimus is the 40-O-(2-hydroxyethyl) derivative of rapamycin [43, 44]. Both everolimus and rapamycin have a similar target affinity, antitumor potency, and spectrum of activity [45–49]. Although both drugs are relatively lipophilic and can readily cross the blood–brain barrier, everolimus has improved CNS penetration compared with rapamycin [46]. Moreover, everolimus has greater water solubility and greater bioavailability [47–49], whereas rapamycin is practically insoluble in water [47, 48]. The systemic bioavailability of everolimus, estimated by the ratio of dose-normalized blood AUCs, amounted to >16 % compared with 10 % for rapamycin [49]. Another mTOR inhibitor, temsirolimus (Torisel), also a derivative of rapamycin, has been shown to be suitable for intravenous administration [7]. Ridaforolimus, available as both an oral and parenteral formulation, is also an mTOR inhibitor, but it has been less extensively studied [7].
6.4.1 Neurological Manifestations
6.4.1.1 SEGAs
Preclinical Studies
Early preclinical studies showed that homozygous loss of either TSC1 or TSC2 results in an embryonically lethal phenotype in mice [50–52]. However, subsequent developments with mouse embryonic fibroblast (MEF) cell lines derived from TSC-null mice and conditional knockout mice yielded important insights on the TSC/mTOR signaling axis. TSC2 −/− MEFs exhibited rapid growth as compared with control MEFs but were strikingly susceptible to inhibition of proliferation by rapamycin [53]. These TSC2 −/− MEFs also expressed high levels of phospho-S6K and phospho-4E-BP1 [53]. Akin to the inhibitory effect of rapamycin on proliferation, rapamycin treatment also induced dephosphorylation of phospho-S6K and phospho-4E-BP1 in order to restrict cell proliferation. This preliminary evidence from murine models suggested that an mTOR inhibitor-mediated blockade of mTORC1 could lead to the suppression of uncontrolled cell proliferation as seen in TSC.
Case Reports
An initial report by Franz et al., wherein clinical improvement in patients with TSC-associated SEGA was noted upon treatment with rapamycin, became the foundation of clinical investigation of mTOR inhibitors in patients with TSC [54]. In a small series of five patients, oral treatment with rapamycin resulted in regression of brain lesions in all the patients (Fig. 6.1). In fact, interruption of treatment in one patient led to SEGA regrowth but showed regression upon readministration of rapamycin [54]. In a subsequent case study, treatment of a 21-year-old woman with oral rapamycin for 5 months resulted in regression of bilateral SEGAs [55]. Lam et al. reported a 50–65 % decrease in SEGAs during 3-month follow-up magnetic resonance imaging (MRI) in three pediatric patients with TSC who received oral rapamycin [56]. Treatment of a patient with TSC-associated SEGA with everolimus for 11 months resulted in significant regression of the lesion and improvement in vision. Unlike previous reports where tumor regrowth occurred upon cessation of rapamycin therapy, in this patient, the SEGAs remained stable and did not show any regrowth during the 9 months following interruption of everolimus therapy [57]. In another case study, treatment of an 11-year-old boy with everolimus at a dose of 4.5 mg/m2/day resulted in several dramatic improvements [58]. After 4 months of everolimus treatment, there was a ≥50 % reduction in the tumor volume. Twelve months after initiation of everolimus therapy, the tumor size was stable and the patient was seizure-free. Taken together, these independent case reports demonstrate the effectiveness of mTOR inhibitors in SEGA and other neurological pathologies associated with TSC.
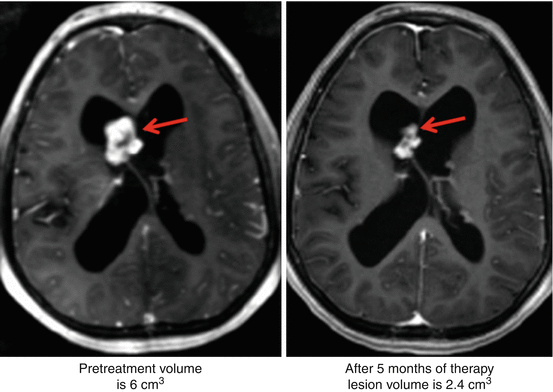
Fig. 6.1
Case report: Reduction in size of SEGA after 5 months of rapamycin therapy. (Left) Pretreatment: lesion volume is 6 cm3. (Right) After treatment: lesion volume is 2.4 cm3. Axial T1 contrast-enhanced MRI. Red arrows point to the SEGA lesion which has shrunk in the right side compared to the left (Reprinted with permission from Franz et al. [54])
Clinical Trials
The observations from the early case reports formed the basis for the phase 2 evaluation of everolimus for the treatment of TSC-associated SEGAs.
Phase 2 Clinical Trial
In the phase 2 study C2485, 28 eligible patients (17 males and 11 females) with TSC-associated SEGAs were enrolled [59]. The initial starting dose of everolimus in the trial was 3.0 mg/m2/day, subsequently adjusted to attain whole-blood trough levels of 5–15 ng/mL. Everolimus therapy was associated with a clinically meaningful and statistically significant reduction in the mean SEGA volume from 2.45 cm3 at baseline to 1.30 cm3 at 6 months (central review; P < 0.001). Of 28 evaluable patients, 21 (75 %) showed at least a 30 % reduction and 9 (32 %) experienced at least a 50 % reduction in the SEGA volume. Furthermore, everolimus therapy was associated with a clinically relevant reduction in the overall frequency of clinical and subclinical seizures (median change, −1; P = 0.02). Quality of life as assessed by QOLCE (Quality-of-Life in Childhood Epilepsy) questionnaire also showed improvement over time. The mean (±SD) scores were 57.8 ± 14 at baseline, 63.4 ± 12.4 at 3 months, and 62.1 ± 14.2 at 6 months. Interestingly, 13 of 15 patients also exhibited an improvement in facial angiofibromas at 6 months of everolimus treatment. All patients had at least one adverse event (AE), which were generally grade 1 (mild) or grade 2 (moderate). The most commonly reported AEs were self-limited infections, primarily upper respiratory infections (79 %) and stomatitis (79 %). Recently published, 3-year, long-term efficacy and safety data showed that at all time points (18, 24, 30, and 36 months), primary SEGA volume were reduced by ≥30 % from the baseline in 65–79 % of patients [60]. The most common AEs were the same as what was reported during the core phase, with no new events in the extension phase. The trial is currently in its fourth year of extension to assess the long-term safety and efficacy of everolimus.
In a subgroup analysis to evaluate the effect of everolimus on normal-appearing white matter (NAWM) using diffusion tensor imaging (DTI), everolimus treatment was associated with a significant increase in fractional diffusivity in corpus callosum, internal capsule, and geniculocalcarine tracts at follow-up at 12–18 months [61]. Moreover, radial diffusivity decreased significantly in corpus callosum and geniculocalcarine tracts, whereas mean total diffusivity decreased significantly only in corpus callosum. The magnitude of changes was small yet statistically significant, demonstrating the ability of pharmacotherapy to modify the genetic defect of TSC in the brain, even in patients with NAWM.
Results from the trial led to the US and EMA (European Medicines Agency) approval of everolimus for TSC-associated SEGAs in pediatric and adult patients with TSC who have SEGAs that require therapeutic intervention but cannot be curatively resected [62].
Phase 3 Clinical Trial
The data from the phase 2 trial formed the basis for the placebo-controlled, multicenter phase 3 trial, EXIST-1(EXamining Everolimus In a Study of Tuberous Sclerosis Complex-1) [63]. A total of 117 eligible patients were randomized in a 2:1 ratio to receive everolimus (n = 78) or matching placebo (n = 39), stratified according to the use of enzyme-inducing antiepileptic drugs (EIAEDs). Everolimus was administered orally at a starting dose of 4.5 mg/m2 of body surface area per day and subsequently adjusted to attain blood trough levels of 5–15 ng/mL. The primary study end point was the proportion of patients with confirmed SEGA response defined as reduction in sum of volumes of all target lesions ≥50 % relative to baseline in the absence of a nontarget lesion worsening, new lesions ≥1 cm in diameter, or new/worsening hydrocephalus. The best overall SEGA response rate was significantly greater in the everolimus group (35 %; 95 % CI, 24.2–46.2) vs. placebo (0 %; 95 % CI, 0.0–9.0). Key secondary and exploratory end points included change from baseline in seizure frequency at 6 months, median time to SEGA progression, skin lesion response rate, and reduction in renal angiomyolipoma volume. The median change from baseline in seizure frequency at week 24 was 0 for both everolimus and placebo arms (P = 0.2004) in contrast to the phase 2 trial, which showed a statistically significant reduction in seizure frequency. The median number of seizures in both treatment arms was 0 at baseline and thus the analysis was inconclusive. One hundred ten patients had at least one skin lesion at baseline. At 24 weeks, 30 (42 %) of 72 patients in the everolimus arm and 4 (11 %) of 38 in the placebo group had a skin lesion response (partial; P = 0.0004). Median time to SEGA progression was not reached in either the everolimus or placebo arm. Interestingly, of 44 patients who had at least one renal angiomyolipoma at baseline (30 in the everolimus arm and 14 in the placebo arm), 16 (53 %) in the everolimus arm had an angiomyolipoma response compared with 0 in the placebo arm. The AE profile was consistent with the known safety profile of everolimus. Most AEs were grade 1 or 2. The most common AEs reported in ≥15 % of patients were mouth ulceration, stomatitis, convulsion, pyrexia, vomiting, nasopharyngitis, and upper respiratory tract infection. In females aged ≥13 years, three out of eight in the everolimus arm developed secondary amenorrhea compared with 0 in the placebo arm. Two cases resolved without intervention, and one resolved with progesterone. Results from this trial clearly showed the clinically meaningful benefit of everolimus with respect to the reductions in TSC-associated SEGA volume. The results from the phase 2 and EXIST-1 trials are summarized in Table 6.2.
Table 6.2
Clinical trials with everolimus in individuals with TSC
Trial | T patients | N | Study design | Treatment | Outcome |
|---|---|---|---|---|---|
C2485 Krueger et al. [59] | Patients with serial growth of TSC-associated SEGA | 28 | Open label, phase 1/2 | Everolimus 3.0 mg/m2/daily Blood trough level: 5–15 ng/mL | Primary end point Reduction in mean SEGA volume at 6 months vs. baseline Secondary end point Reduction in the overall frequency of clinical and subclinical seizures Improvement in quality of life Improvement in facial angiofibromas |
EXIST-1 Franz et al. [63] | Patients with serial growth of TSC-associated SEGA | 117 | Randomized, double blind, placebo controlled, phase 3 | Everolimus vs. placebo 4.5 mg/m2 of body surface area/daily Blood trough level: 5–15 ng/mL | Primary end point SEGA volume response (≥50 % reduction from baseline): everolimus 35 % vs. placebo 0 % Secondary end point Seizure frequency: no difference in mean change from baseline between everolimus and placebo groups Skin lesion response: everolimus 42 % vs. placebo 11 % Renal angiomyolipoma response: everolimus 53 % vs. placebo 0 % |
EXIST-2 Bissler et al. [64] | Patients with TSC- or sporadic LAM-associated angiomyolipoma | 118 | Randomized, double blind, placebo controlled, phase 3 | Everolimus vs. placebo 10 mg once/daily | Primary end point Renal angiomyolipoma response rate (≥50 % reduction in volume from baseline): everolimus 42 % vs. placebo 0 % Secondary end point Skin lesion response rate: everolimus 26 % vs. placebo 0 % Lung function: limited analysis due to short duration of treatment and low number of patients |
6.4.1.2 TSC-Associated Epilepsy and Neurocognition Impairment
Preclinical Studies
Effects of mTOR hyperactivation on developmental and functional abnormalities in the brain and the role of mTOR inhibition have been extensively studied in mouse models with conditional inactivation of TSC1 or TSC2 genes. Treatment of TSC1 GFAP CKO mice, which have conditional inactivation of TSC1 in glial fibrillary acidic protein (GFAP)-positive cells, with rapamycin, blocked the activation of mTOR pathway, prevented astrogliosis in both neocortex and hippocampus, prevented the brain enlargement typically seen in these mice, and restored the compact organization of the pyramidal cell layer of hippocampus. Early treatment of TSC1 GFAP CKO mice with rapamycin also prevented development of seizures and increased the survival of these mice. Similarly, treatment of TSC2 GFAP CKO mice with rapamycin also resulted in reversal of the neurological phenotype [65]. In a conditional neuronal mouse model of TSC, mice developed several TSC-related neuropathologies such as enlarged and dysplastic neurons, reduced myelination, and high expression of phospho-S6, as well as a poor median survival of 33 days. Treatment of these mice with either rapamycin or everolimus resulted in an overall increased survival rate with a median of 80 days [66]. In neuronal subset-Pten (NS-Pten) mouse model of cortical dysplasia, mice exhibited hypertrophic neurons, spontaneous seizures, abnormal EEG activity, as well as increased activation of mTOR pathway, all of which was suppressed by rapamycin treatment [67]. Mice with mosaic induction of TSC1 loss in neural progenitor cells displayed multiple neurological symptoms such as severe epilepsy and premature death. Postnatal treatment reversed the neurological phenotype of these mice and rescued the mice from epilepsy and premature death [68]. In a rat model of temporal lobe epilepsy in which kainate-induced seizures resulted in biphasic hyperactivation of mTOR, treatment with rapamycin blocked both the acute and chronic phases of seizure-induced mTOR activation and decreased development of spontaneous epilepsy [69]. Learning and memory deficits seen in TSC2 +/− mice were shown to be ameliorated by brief treatment with rapamycin, demonstrating the potential of mTOR inhibition in the treatment of neurocognitive and behavioral manifestations of TSC [70].
Case Reports
Muncy et al. reported the first trial of oral rapamycin for seizures in a 10-year-old girl with TSC who continued to have seizure clusters even after resection of two cortical tubers identified as primary areas of seizures. At a rapamycin dose of 0.15 mg/kg/d (level 9.8 ng/mL), seizure clusters completely stopped, although pre- and posttreatment MRI did not show any change in the cortical tubers. Perek-Poinik et al. also reported cessation of intractable seizures in a critically ill 10-year-old boy with TSC upon treatment with everolimus (Sect. 6.4.1.1) [58].
Clinical Trials
Krueger et al. recently published the first prospective human trial to assess whether everolimus could also benefit patients with epilepsy and TSC [71]. In this multicenter, open-label, phase 1/2 clinical trial, 20 of the 23 enrolled patients with medically refractory epilepsy were treated with everolimus for a total of 12 weeks. Seizure frequency was reduced by ≥50 % in 12 of 20 subjects. Overall, the median seizure frequency was decreased by 73 %. Four subjects (20 %) were free of clinical seizures and seven (35 %) had at least a 90 % reduction seizure frequency. Although the sample size was small in the study, the effect of everolimus was antiepileptogenic rather than conventional anticonvulsant, since these patients had failed at least two AED regimens. Moreover, the AEs were mild or moderate in severity. Effect of everolimus on epilepsy has also been studied as a secondary end point in two separate clinical trials that yielded conflicting results [59, 63]. In the phase 2 trial of everolimus for TSC-associated SEGA, everolimus therapy was associated with a statistically significant reduction in the overall frequency of seizures in 9 out of 16 patients for whom video-EEG data were available (Sect. 6.4.1.1) [59]. However, these findings could not be duplicated in the subsequent phase 3 trial (EXIST-1) that involved 117 patients with TSC-associated SEGA [63]. A large, randomized, double-blind, placebo-controlled study (EXIST-3) to evaluate the efficacy and safety of everolimus as an adjunctive therapy in patients with TSC who have refractory partial-onset seizures is currently ongoing.
6.4.2 TSC-Associated Angiomyolipomas
6.4.2.1 Preclinical Studies
TSC mouse models of kidney lesions have been used to study the effect of mTOR inhibitors on renal tumor size and growth. TSC2 +/− mice that develop spontaneous kidney cystadenomas at high frequency by the age of 12 months show dramatic reduction in the severity of kidney disease upon treatment with CCI-779 (a rapamycin analog) [72]. Moreover, treatment of nude mice harboring TSC2 −/− tumors with CCI-779 resulted in reduced tumor growth and improved survival compared with untreated nude mice [72]. In an ENU-accelerated TSC2 +/− tumor model, treatment with everolimus was shown to be highly effective in reducing the gross tumor size, microscopic tumor size, and percent solid tumor [73]. Everolimus therapy correlated with a marked reduction in expression of pS6, consistent with blockade of mTOR signaling. However, brisk tumor growth resumed when the mice were taken off the drug. In an Eker rat model of TSC-bearing germline TSC2 mutation, rapamycin resulted in downregulation of mTOR activity in renal tumors [74]. In addition, rapamycin reduced the size of TSC2-related renal tumors. Taken together, these data provide strong preclinical evidence for the role of mTOR inhibitors in the treatment of renal disease associated with TSC.
Related posts:
 The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
 Predictive Biomarkers of Response to mTOR Inhibitors
Predictive Biomarkers of Response to mTOR Inhibitors
 mTOR Inhibition Beyond Rapalogs
mTOR Inhibition Beyond Rapalogs
 Rational Combinations of mTOR Inhibitors as Anticancer Strategies
Rational Combinations of mTOR Inhibitors as Anticancer Strategies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


