Radionuclide
T 1/2 (h)a
Emissionsb
Emax (keV)c
Range max in soft tissue (mm)c
Usual labeling method
Technetium-99 m
6.0
γ
140
Direct labeling or N2S2 or N3S complexes
Indium-111
67
γ
171 and 245
Polyamino-carboxylic acids: DTPA, DOTA
Iodine-123
13.3
γ
159
Direct labeling (tyrosine)
Fluorine-18
1.83
β+
633
3.1
Gallium-68
1.13
β+
1,899
9.8
Polyamino-carboxylic acids: DOTA, NOTA
Copper-64
12.7
β+
653
3.2
Many different chelating agents
β−
579
2.8
Zirconium-89
78
β+
902
4.6
Desferroxamine
Iodine-124
100
β+
1,535 and 2,138
7.9 and 10.9
Direct labeling (tyrosine)
Scandium-44
3.97
β+
1,473
7.6
Iodine-131
193
β−
610
2.9
Direct labeling (tyrosine)
γ
362
11
Yttrium-90
64
β−
2,250
11
Polyamino-carboxylic acids: DOTA
Rhenium-188
17
β−
2,120
10
Direct labeling or N2S2 or N3S complexes (chemistry analogous to that of technetium)
γ
155
Lutetium-177
162
β−
498
2.0
Polyamino-carboxylic acids: DOTA
γ
208
Copper-67
62
β−
392–577
1.8
Many different chelating agents
γ
184
Bismuth-213
0.76
α
8,400
0.1
Polyamino-carboxylic acids: CHX-DTPA, DOTA
γ
440
Astatine-211
7.2
α
5,870 and 7,450
0.055–0.080
Stannylated synthons: SAB, SAPS
X
77–92
For alpha emitters, 213Bi is available through a generator made of 225Ac. Its short half-life makes it tricky to use; nonetheless, many studies are ongoing worldwide [22]. Despite its complex chemistry, 211At may be a better candidate for alpha therapy due to its longer half-life and its production in accelerators. Other alpha emitters are available but they are linked to a cascade of alpha decays that may be a problem for specific targeting (225Ac and 226Th) or have a chemistry not favorable for labeling (223Ra).
Regarding the targeting aspect, the specificity of the radiopharmaceutical has to be the highest possible to limit the delivery of radionuclides to healthy tissues, and affinity controls the uptake of the radiopharmaceutical in target lesions: higher affinity means higher uptake, although affinities in the nanomolar range are considered sufficient. For longer half-life radionuclides, the rate of efflux from the tumor is also very important and is not entirely related to affinity. For instance, when internalization of the radiopharmaceutical by target cells occurs, residualizing radionuclides, such as metals, affords protracted radioactivity retention in tumor sites, whereas direct radiolabeling with radioiodine results in fast excretion of radioactivity, thus reducing target cell exposure.
25.3.2 Labeling Techniques
Halogens are usually provided under the halogenide form. The easiest way to attach a halogen to a vector is to perform an electrophilic substitution on a tyrosine residue [23]. Unfortunately, even if this commonly used technology is validated with iodine-labeled non-internalizing antibodies or peptides, it does not provide satisfactory results when internalization occurs or with 211At-labeled antibodies [24]. In both cases, halogen liberation leads to nonspecific irradiation of normal organs such as the thyroid for iodine [25] or the stomach for astatine [26] and reduces specific irradiation of the tumor. Several radiolabeling approaches using prosthetic groups have been proposed to solve this problem (Fig. 25.1).
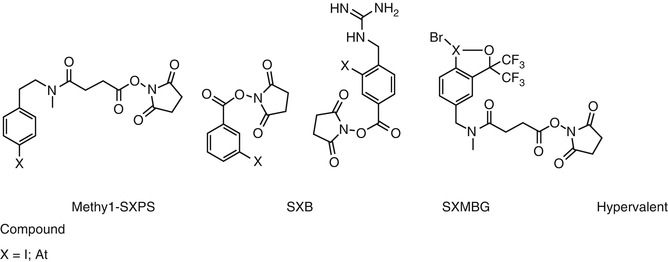
Fig. 25.1
Prosthetic groups for radiohalogen labeling
Radiometals, such as 90Y, 188Re, 67Cu, 177Lu, 212Bi, and other actinides, are generally provided no carrier added in chloride form. However, contaminations with metal traces resulting from the production mode decrease the specific activity of radiopharmaceuticals, which are usually between 40 and 400 MBq/nmol depending on the radionuclide. Several highly specific chelating agents have been developed in order to improve specific activity [27].
Transmetallation or transchelation phenomena can occur in vivo when the radiopharmaceutical is in competition with metal complexing proteins, such as transferrin or ceruloplasmin [28, 29]. Thus, chelating agents with very high affinities for metals and very high kinetic stabilities have been developed (Table 25.1). The best approaches to limit these phenomena are based on a better chelation agent selection in order to improve both selectivity and stability. This choice integrates stability constant and dissociation kinetic values which have to be for the latter as low as possible.
25.4 The Treatment of B Cell Lymphoma with Anti-CD20 Antibodies
Bexxar® and Zevalin® are administered 6–8 days after a predose of cold mAbs, respectively 2 × 450 mg of tositumomab and 2 × 250 mg of rituximab, to improve biodistribution and tumor targeting. Bexxar® and Zevalin® can be integrated in clinical practice using non-ablative doses for treatment of patients with relapsed or refractory FL or as consolidation after induction chemotherapy in frontline treatment in FL patients. Hematological toxicity is the major side effect of RIT and depends on bone marrow involvement and prior treatment [30–32]. Non-hematological toxicity is generally low. Secondary myelodysplastic syndrome or acute myelogenous leukemia (AML) was reported in 1–3 % of cases [30–33]. The risk appears to be increased in patients previously treated by several lines of chemotherapy or radiotherapy. In a meta-analysis involving relapsed B cell lymphoma patients treated with Zevalin® in four clinical trials, long-term responses (time to progression (TTP) >12 months) were seen in 37 % of patients [32]. At a median follow-up time of 53.5 months, the median TTP was 29.3 months. One third of these patients had been treated with at least three previous therapies, and 37 % of them had not responded to their last therapy. The estimated 5-year overall survival (OS) was 53 % for all patients treated with Zevalin® and 81 % for long-term responders. Using Bexxar® in a long-term meta-analysis performed on 250 heavily pretreated patients with indolent lymphoma in 5 clinical trials, objective response (OR) rates ranged from 47 to 68 % and complete response (CR) rates from 20 to 38 % [34]. Interestingly, poor prognostic patients showed durable responses (bone marrow involvement in 41 %, bulky disease ≥5 cm in 49 %, and transformed histology in 23 %).
Clinical results showed that Zevalin® or Bexxar® had a significant efficacy but moderate response duration as a monotherapy in rituximab-refractory recurrence of FL. A higher therapeutic impact may be achieved using Bexxar® or Zevalin® in other indications. Recent studies showed that RIT can be administrated as high-dose treatment. This approach consists of injecting myeloablative activity of RIT or combining standard or escalated activity of RIT with high-dose chemotherapy. In a recent prospective multicenter study, Shimoni et al. demonstrated that standard-dose Zevalin® (0.4 mCi/kg) combined with BEAM high-dose chemotherapy was safe and possibly more effective than BEAM alone as a conditioning regimen for stem cell transplantation (SCT) in 43 patients with relapsed/refractory aggressive non-Hodgkin lymphoma [35]. The 2-year progression-free survival (PFS) was 59 and 37 % in the Z- BEAM and BEAM arms, and the 2-year OS was 91 and 62 %, respectively.
RIT can also be administered as consolidation after induction therapy. The FIT randomized phase III trial showed the benefits of Zevalin® as consolidation in previously untreated FL patients [7]. A high conversion rate from partial response (PR) to CR of 77 % was observed after RIT, leading to a high CR rate of 87 %. Moreover, different studies suggest that RIT is a relevant option as consolidation therapy in different subtypes of B cell lymphoma such as diffuse large B cell or mantle cell lymphoma, in order to decrease the number of chemotherapy courses in elderly patients or as an alternative of stem cell transplantation in high-risk patients [36, 37]. In 2010, Zinzani et al. published the results of a phase II study assessing the efficacy and safety of Zevalin® following four cycles of R-CHOP21, in 55 high-risk elderly (age ≥60 years) patients with previously untreated diffuse large B cell lymphoma (DLBCL). Forty-eight of the 55 patients received RIT [38]. The OR rate for the entire treatment regimen was 80 %, including 73 % CR. Eight of the 16 patients (50 %) who achieved less than a CR after R-CHOP improved their remission status after RIT. With a median follow-up of 18 months, the 2-year PFS was estimated to be 85 %, with a 2-year OS of 86 %.
RIT can also be considered alone in frontline treatment. Recently, Scholz et al. evaluated, in an international multicenter phase II clinical trial, the efficacy and feasibility of Zevalin® as first-line treatment in 59 FL patients [39]. Treatment indication resulted from B symptoms, grade 3A, organ compression or infiltration, rapid growth, and/or bulky disease. The OR rate at 6 months after RIT was 87 %, with 41 % of the patients achieving CR, 15 % unconfirmed CR, and 31 % PR. Median PFS was 25.9 months. RIT was well tolerated and the most common toxicity was hematological and reversible.
25.5 Promising Results in Hemopathies Using Other Antibodies
25.5.1 Targeting of Lymphoma with Anti-CD22 Antibodies
For lymphoma, targeting other antigens than CD20 targeted by rituximab appears relevant, offering the possibility of targeting populations of cells not expressing CD20 or not responding to cold anti-CD20 mAbs. CD22 is a transmembrane glycoprotein expressed on mature B cells but not expressed on stem cells or plasma cells and functions in B-cell regulation/activation. CD22 is highly expressed across malignant B-cell histologies. The anti-CD22 epratuzumab has good features for RIT because it is humanized, internalized by target cells, stably labeled using DOTA, and administered without a loading dose of cold antibody, at variance with Zevalin® or Bexxar® [40].
90Y-epratuzumab RIT has been developed with repeated injections [41–43]. A multicenter phase I/II study was designed to assess fractionated 90Y-epratuzumab in NHL relapsing patients [43]. Sixty-four patients with one to five prior therapies (median, 2), with different histologies of B cell lymphoma were enrolled. The total 90Y activities ranged from 0.185 to 1.665 GBq/m2, with comparable numbers treated at ≤0.37 (N = 17), > 0.37–0.74 (N = 13), > 0.74–1.11 (N = 16), and >1.11 GBq/m2 (N = 18). Even at the highest total 90Y activity of 1.665 GBq/m2, grade 3–4 hematological toxicities were manageable with support in patients with <25 % bone marrow involvement. The overall OR rate was 62 % (48 % CR/unconfirmed CR). For FL patients without prior SCT, response rates increased with total 90Y activity, with 92 % CR/unconfirmed CR at the highest dose levels (>1.11 GBq/m2). Patients with CR/unconfirmed CR achieved long-lived responses continuing up to 5 years, including 24.6-month median PFS for 12 FL patients receiving >1.11 GBq/m2 total 90Y activity.
Targeting of antigens other than CD20 appears particularly interesting in the context of consolidation therapy after rituximab-based therapy. A French phase II trial sponsored by the LYSA group is ongoing assessing frontline treatment using fractionated RIT with 90Y-epratuzumab as consolidation therapy after chemo-immunotherapy in bulky or stage III/IV aggressive B cell lymphoma. Another important perspective is the clinical evaluation of dual-targeted antibody/radioantibody therapy [40, 44, 45]. Combining an unconjugated anti-CD20 antibody therapy with a radioimmunoconjugate binding to a noncompeting antigen might improve responses by allowing optimal uptake of each agent [45, 46]. Preclinical studies showed that efficacy increased when a consolidation using anti-CD20 veltuzumab was delivered after anti-CD22 RIT [46]. The injection of cold mAb after the radioactivity dose provided higher efficacy than injection before RIT, and the amount of predose of cold mAb could be minimized [40, 45]. Thus, a reexamination of RIT in the treatment of B cell lymphoma was proposed [44], emphasizing that in RIT clinical practice, nearly 900 mg of unlabeled anti-CD20 IgG antibody is predosed to the patient before the anti-CD20 90Y or 131I RIT.
25.5.2 Targeting of Multiple Myeloma Using Anti-CD138 Antibodies
Multiple myeloma (MM) is a malignant plasma cell disorder characterized by the proliferation of clonal cells in the bone marrow and at later stages of the disease in extramedullary sites [47]. The annual incidence is 4–6 cases per 100,000. The median survival of this incurable disease has markedly improved over the last decade due to the extensive use of high-dose therapy and autologous stem cell transplantation in younger patients and to the broad introduction of novel agents, i.e., thalidomide, bortezomib, and lenalidomide, used in combination with dexamethasone or alkylating agents [48]. Other drugs such as inhibitors of histone deacetylase (vorinostat, panobinostat) or mAbs (elotuzumab) are under development in large prospective phase II or III studies [49].
Numerous immunotherapy approaches targeting MM cell surface antigens have been tested. Preclinical and clinical trials have been conducted with naked mAbs having an intrinsic cytotoxic action, interfering with ligand binding or involved in angiogenesis. Anti-CD20 rituximab [50], anti-CD38 [51], anti-CD54 [52], anti-CD74 [53], anti-CD317 [54, 55], or anti-CD319 [56] has been assessed as monotherapy or in combination with other therapeutic drugs or in preparation of autologous SCT. Because IL-6 is a major autocrine/paracrine growth factor for MM cells, immunotherapy with anti-IL-6 mAb has been performed. A transient tumor cytostasis was obtained, which did not cure the tumor [57, 58]. Finally, Lee et al. have shown the expression of CD66a but not of other CD66 isoforms in MM. These findings open the possibility of using mAbs against members of the carcinoembryonic antigen (CEA) and immunoglobulin superfamily in RIT [59]. Erba et al. have performed a RIT clinical trial using 131I-L19SIP mAb specific to the EDB domain of fibronectin, reporting a stabilization of the disease in two patients at advanced stage of MM [60]. The feasibility of anti-CD138 (syndecan-1) RIT using 131I-B-B4 was also recently reported, with encouraging dosimetry results [61]. Syndecan-1 belongs to the family of heparan sulfate-bearing proteoglycans. Found on epitheliums, this molecule is also present on pre-B cells and plasma cells, and it plays an important role in regulating MM [62]. Syndecan-1 is expressed in all MM tumors within the bone marrow and is present at relatively high levels on MM cell surface [62–65].
In MM, tumor cells are mostly disseminated in bone marrow either as isolated cells or as microscopic tumor cell clusters. Beta emitters with relatively long path lengths (1 mm–1 cm) are not very suitable to target such isolated cells. By contrast, the high linear energy transfer characteristics of alpha particles enable localized irradiations while preserving surrounding tissues, and cell toxicity is achieved with only a few disintegrations at the cell surface. In vitro and preclinical studies demonstrated promising therapeutic efficacy of 213Bi-labeled anti-mCD138 for the treatment of MM [66]. CD138 targeting with a mAb coupled to a radionuclide emitting alpha particles thus represents a potential new therapeutic option for MM, and the use of alpha emitters with longer half-lives, such as 211At (7.2 h), should be evaluated in the clinic.
25.6 RIT of Metastatic Prostate Cancer
PCa accounts for an estimated 70,347 deaths in Europe in 2013 [67]. Up to 40 % of patients eventually develop metastases despite local therapy. Once metastases have developed, PCa is incurable and all therapy is palliative. Medical castration is highly effective in shrinking tumor burden, decreasing prostate-specific antigen (PSA) levels, enhancing quality of life, and improving survival [68]. However, most patients evolve toward progression despite castration, with a median duration of response of 12–24 months [68]. At the stage of castration-resistant PCa (CRPC), cytotoxic chemotherapy was the only therapy [69, 70] until 2012, when the European Medicines Agency (EMA) approved the use at this stage of abiraterone acetate before docetaxel. Within the past year, three new drugs were FDA approved for the treatment of patients with CRPC (cabazitaxel, sipuleucel-T, and denosumab). However, the survival benefit of these drugs in CRPC is modest: respectively +2.4, +4.1, and +3.6 months, and more efficacious drugs are needed.
Radiotherapy is an established treatment for clinically localized PCa or for palliation of painful bone metastasis [71]. PCa is a favorable solid malignancy for which RIT may be used because it is a radiosensitive tumor with typical distribution to sites with high exposure to circulating radiolabeled mAbs (bone marrow and lymph nodes). In preclinical and clinical PCa therapy studies, radionuclides have been linked to antibodies or peptides with affinity to mucin, ganglioside (L6), Lewis Y (Ley), adenocarcinoma-associated antigens, and prostate-specific membrane antigen (PSMA) [72–75], but PSMA appears the most specific.
PSMA is an integral, non-secreted, type II membrane protein with abundant and nearly universal expression on prostate epithelial cells and is strongly upregulated in PCa [76–80]. Pathology studies indicate that PSMA is expressed by virtually all PCa [81]. The level of expression in non-prostate tissues is 100–1,000-fold less than in prostate tissue [76], and the site of PSMA expression in normal cells (brush border/luminal location) is not typically exposed to circulating mAb. De-immunized J591 mAb, which targets the external domain of PSMA, giving an easy and rapid access to the antigen, seems to be the best clinical candidate for imaging and therapy of PCa [82, 83].
A phase I trial assessing 111In/90Y-J591 was performed in 29 patients [84]. Dose-limiting toxicity was seen at 740 MBq/m2, and 647.5 MBq/m2 was determined as the maximal tolerated dose (MTD). The overall targeting sensitivity of bone and soft tissue metastasis was 81 %. Decrease of PSA was observed for two patients as objective measurable disease responses with decrease of lymph node size.
Thirty-five patients were enrolled in a 177Lu-J591 phase I trial [85]. The 2,590 MBq/m2 level was determined as MTD. Repeated dosing up to three doses of 1,110 MBq/m2 could be safely administered. Clearly identified sites of metastatic disease were successfully imaged by 177Lu-J591 scintigraphy in 100 % of patients. The median duration of PSA stabilization, after treatment, was 60 days with a range of 28–601 days. No immune response was detected. A phase II 177Lu-J591 trial was initiated in CRPC patients (ASCO congress 2008). Fifteen patients (cohort 1) were treated with 2,405 MBq/m2. The second cohort (2,590 MBq/m2) enrolled 17 patients (ASCO congress 2013). Sensitivity of known metastasis targeting was 93.6 %. Reversible thrombocytopenia and neutropenia toxicity occurred respectively in 46.8 and 25.5 %. The second cohort dose (2,590 MBq/m2) showed more PSA responses (46.9 % vs. 13.3 %, p = 0.048) associated with a longer survival (21.8 vs. 11.9 months, p = 0.03) but also more reversible hematological toxicity.
These trials provide support that radiolabeled de-immunized J591 is well tolerated and non-immunogenic. Radiolabeled J591 effectively targets PCa metastases with high sensitivity and specificity, produces PSA, and declines with a dose-effect relationship.
25.7 RIT with Alpha-Emitting Radionuclides
Alpha-RIT is a therapeutic modality based on the use of an antitumor antigen mAbs coupled to an alpha emitter (radionuclide which decays by the emission of alpha particles). The rationale of this therapeutic modality is based on two prominent characteristics of the alpha particles: their short range in tissue, inferior to 100 μm, which allows for a good specificity of the treatment (once the antibody is in the vicinity of the tumor) and their high linear energy transfer (LET) between 50 and 250 keV/μm, which makes them highly cytotoxic. In vitro studies have demonstrated that 1–20 cell nucleus traversals by alpha particles are sufficient to inactivate a cell as compared to thousands or tens of thousands for the same effect with beta− particles [86, 87]. In addition, alpha particle-induced toxicity was shown to be independent of both dose rate and oxygenation of the irradiated tissue [88].
25.7.1 Therapeutic Indication
Related to these characteristics, it is often described that alpha-RIT is particularly indicated in the treatment of MRD, hematologic cancers, and micrometastatic diseases, even though some efficacy was observed on solid tumors [22]. Despite trials using alpha-emitting radionuclides in medicine in the early twentieth century, just after the discovery of radioactivity, the first alpha-RIT clinical trial was performed in 1997. A humanized antibody specific for a human myelogenous leukemia antigen (CD33) labeled with 213Bi was administered to 18 patients with AML, and results showed a reduction in circulating blasts in most patients (~80 %), whereas no extramedullary toxicity was observed [89]. Since then, seven clinical trials (Table 25.2) were initiated to treat lymphoma, melanoma [90, 91], malignant recurrent gliomas [14], and ovarian carcinoma [92]. These studies demonstrated that alpha-RIT is feasible and safe and has a significant antitumor efficacy. In addition to these alpha-RIT trials, two clinical trials using alpha-emitting radionuclides against gliomas [93] and against bone metastases in CRPC [94, 95] should be mentioned. In the last indication, the ALSYMPCA trial (phase III) using 223RaCl2 demonstrated a significant prolongation of OS as compared with placebo and the product, Alpharadin, is now on fast track to FDA approval.
Cancer type | Radioconjugate | Phase | No. of patients | Reference |
|---|---|---|---|---|
Leukemia | 213Bi-HuM195mAb | I | 18 | [89] |
213Bi-HuM195mAb | I/II | 31 | [98] | |
225Ac-HuM195mAb | I | Not yet completed | [101] | |
Lymphoma | 213Bi-rituximab | I | 12 | [157] |
Melanoma | 213Bi-9.2.27mAb | I (intralesional) | 16 | [90] |
213Bi-9.2.27mAb | I (systemic) | 38 | [91] | |
Recurrent malignant brain tumors | 211At-81C6mAb | I | 18 | [14] |
Ovarian carcinoma | 211At-MX35mAb | I | 9 | [92] |
25.7.2 Limited Availability
These encouraging results of alpha-RIT in clinics are still limited if we consider the number of patients treated with alpha emitters up to now (n = 142). More than 100 alpha-emitting radionuclides are known, but once selected for appropriate characteristics, less than 10 have been evaluated [96]. Among them, the most promising ones are astatine-211, bismuth-212, bismuth-213, radium-223, actinium-225, and thorium-227. Current supplies of these radionuclides are based on nuclear weapon or nuclear fuel material reprocessing and cyclotron productions [97]. Concerning 213Bi, only three centers in the world are able to produce 225Ac/213Bi generators, and combining all sources of production, only 100–200 patients could be treated annually. For 211At, the production is also insufficient, since it is currently carried out by less than 10 cyclotrons in the world [96]. In the United States, this issue of availability was clearly identified, and recent analyses emphasized the need to develop new infrastructures for the production of alpha-emitting radionuclides.
25.7.3 Issues and Current Developments
Following the first clinical trial using 213Bi-HuM195, it appeared that the limited number of 213Bi atoms that could be injected was a clear issue to allow for complete remission in patients with large tumor burdens [89]. Different ways of optimization have been proposed to overcome this issue. Therapeutic associations of alpha-RIT with chemotherapy were shown to be efficient in vitro, and a clinical trial was initiated including alpha-RIT after partial cytoreduction with cytarabine [98]. Another solution relies on the use of 225Ac, 227Th, or 223Ra. These radionuclides yield several daughter radionuclides with four or more alpha particle emissions in their decay scheme [99, 100]. The main issue here is to manage the distribution, metabolism, and clearance of the daughters since consecutive decays result in the loss of the chemical bond to the antibody [101]. Different solutions have been proposed. For instance, the 225Ac atomic nanogenerator was described using the 225-mAb that internalizes in target cells, thus trapping the radionuclide and its daughters within the target [101].
As for beta-RIT, pretargeting strategies are currently evaluated in order to enhance the therapeutic window of RIT, i.e., to increase the tumor-to-organ ratio in terms of activity delivery [100]. The faster delivery of activity to tumors may also be an advantage when using short-lived alpha-emitting radionuclides (213Bi, 211At).
Finally, increased efforts are made to better understand the toxicity induced by alpha-RIT. Due to its short range, the distribution of radioimmunoconjugates at the sub-organ scale is expected to be crucial for toxicity. Different dosimetry models of organs (kidneys, bone marrow) are developed to determine the dose distribution following a RIT treatment at the cell scale [102, 103].
25.8 High Efficacy of Pretargeting Approaches in Metastatic Thyroid Carcinoma
Medullary thyroid carcinoma (MTC) represents less than 10 % of all thyroid carcinoma. Prognosis of metastatic disease varies from long- to short-term survival. Among the various prognostic parameters, advanced age, stage of the disease, EORTC prognostic scoring system mutations in the RET oncogene, and association with multiple endocrine neoplasia (MEN) 2B are commonly accepted as prognostic factors [104–108]. Moreover, Barbet et al. demonstrated that calcitonin (Ct) serum level doubling times (DT) was an independent predictor of OS [109]. In this study, all the 41 patients with Ct DT >2 years were still alive at the end of the study 2.9–29.5 years after initial surgery. Eight patients (67 %) with DT between 6 months and 2 years died of the disease 40–189 months after surgery, and all 12 patients with Ct DT <6 months died of the disease 6 months to 13.3 years after initial surgery. Giraudet et al. confirmed the prognostic value of biomarker DT in metastatic MTC [110].
Targeted therapy using multikinase inhibitors can be applied in progressive patients and vandetanib has been approved [111–116]. MTC cells express high amounts of CEA, and anti-CEA radiolabeled mAbs have shown promising results [117, 118]. Pretargeted RIT (pRIT) was developed to improve the tumor-to-normal-tissue ratios and to deliver increased tumor-absorbed doses to relatively radioresistant solid tumors. Pretargeted system involves a first injection of an unlabeled bispecific monoclonal antibody (BsmAb), followed by a second injection of a radiolabeled bivalent hapten peptide [1, 19, 119–121]. Using this system, the radiolabeled bivalent peptide binds avidly to the BsmAb attached to the CEA antigen on the cell surface, whereas non-targeted hapten-peptide in the circulation clears rapidly through the kidneys.
A phase I/II clinical trial was started in 1996 to evaluate pRIT using the murine anti-CEA x anti-indium-DTPA F6 × 734 BsmAb and a bivalent indium-DTPA hapten labeled with iodine-131, in 26 metastatic MTC patients [122–124]. A good tumor targeting was observed. Dose-limiting toxicity was hematological, and maximum tolerated activity was estimated at 1.8 GBq/m2 in this population of patients with high frequency of bone marrow involvement. Some tumor responses were observed, mainly in patients with a small tumor burden and after repeated courses of pRIT. Because of a relatively high hematological toxicity and frequent immune responses, the chimeric hMN-14 × m734 BsmAb was developed and assessed in a prospective phase I study performed in 34 patients with CEA-expressing tumors to determine optimal BsmAb dose, hapten activity, and pretargeting interval [22]. A BsmAb dose of 40 mg/m2 with a pretargeting interval of 5 days appeared to be a good compromise between toxicity and efficacy. HAMA elevation was observed in 8 % of patients and HAHA (human antihuman antibody) in 33 %.
In 2006, OS of the series of 29 MTC patients involved in the two phase I/II pRIT trials was retrospectively compared with that of 39 contemporaneous untreated patients (data collected by the French Endocrine Tumor Group, GTE) [125]. A second objective was to examine whether post-pRIT Ct DT variation was a surrogate marker for survival. Patients with Ct DT <2 years were considered as high-risk patients. This study showed that OS was significantly longer in high-risk treated patients than in high-risk untreated patients (median OS, 110 vs. 61 months; P < 0.030).
Following theses encouraging results, a prospective phase II multicenter pRIT trial was designed in progressive MTC patients with Ct DT shorter than 5 years. Forty-two MTC patients received 40 mg/m2 of hMN-14 × m734 and 1.8 GBq/m2 131I-di- indium-DTPA hapten 4–6 days later [126]. Disease control according RECIST criteria (objective response + stabilization) was observed in 32 patients (76.2 %), including a durable CR of at least 40 months in 1 patient (2.4 %) and durable stable disease (≥6 months) in 31 patients (73.8 %). Tumor uptake assessed by post-pRIT immunoscintigraphy was a significant predictor of response. As previously reported, toxicity was mainly hematological, requiring careful post-RIT blood monitoring. Pre-RIT biomarker DT and impact on DT after pRIT were predictors of OS, confirming the value of serum biomarkers in selecting patients and monitoring therapy.
Today, a new generation of compounds is available for pRIT. Humanized, recombinant, trivalent BsmAb (anti-CEA TF2) and bivalent histamine-succinyl-glutamine (HSG) peptides have been produced [127, 128]. The use of TF2, composed of a humanized anti-HSG Fab fragment derived from the 679 anti-HSG mAb and two humanized anti-CEA Fab fragments derived from the hMN-14 mAb (labetuzumab, Immunomedics, Inc.) by the dock-and-lock procedure, should reduce immunogenicity [127–129]. Moreover, the HSG peptide allows facile and stable labeling with different radiometals, such as 177Lu or 90Y, having favorable physical features that could improve pRIT efficacy [130].
Related posts:
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
 Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


