(a) Diffuse astrocytomas are infiltrative tumors with fibrillar cytoplasm and mildly pleomorphic nuclei.
(b) The cytoplasm of diffuse astrocytomas is generally immunoreactive for glial fibrillary acidic protein, though cell borders are commonly indistinct among the tumor cells.
(c) Mitotic activity is absent in small samples and Ki-67 labeling index is low, usually between 1 and 3%.
The Dumas-Duport grading system18 applies relatively well-defined features in a point system that are useful in distinguishing among grade 2, grade 3, and grade 4 fibrillary astrocytomas. In this system, one point (usually nuclear pleomorphism) equals grade 2, two points (plus mitoses) equals grade 3, and 3 or 4 points (plus vascular proliferation and/or necrosis) equals grade 4. Translating into the WHO system, the absence of frequent mitotic activity, endothelial vascular proliferation and necrosis are diagnostic features of grade II astrocytomas. However, individually necrotic tumor cells may be encountered and should not be confused with mitotic figures. Other degenerative forms, including the formation of microcysts, may be frequent and microcalcification may occasionally be identified.
Astrocytomas are immunoreactive for S100 and GFAP. Immunohistochemistry for the Ki-67 antigen (MIB-1 antibody) reveals a low labeling index that, at Duke, is less than 4% of tumor cell nuclei (Figure 7.1c). Occasionally one may encounter a relatively sparsely cellular glial neoplasm exhibiting a brisk Ki-67 labeling index. In such examples, we raise the possibility of the intensely infiltrative astrocytic variant, gliomatosis cerebri. While approximately 50% of diffuse astrocytomas exhibit mutations of TP53, recent studies indicate that 85% of gliomas (both astrocytomas and oligodendrogliomas) exhibit a recurrent mutation in one of the isocitrate dehydrogenase (IDH) genes, IDH1 or IDH2, most commonly IDH1.19 A monoclonal antibody has been made to the most common mutation and used successfully in characterizing a glial population as neoplastic as well as in distinguishing an astrocytoma from an IDH1 intact pilocytic astrocytoma.20
Anaplastic astrocytoma
Epidemiologic,16 histologic, and molecular research indicates that anaplastic astrocytoma represents an intermediate stage in malignant progression from well-differentiated astrocytoma to secondary glioblastoma (GBM). Grossly, the anaplastic astrocytoma is distinguished amidst its background of low-grade glioma by its friable, gray granular tissue that imperceptibly merges with the surrounding brain. Multifocality of the change can be found; however, the presence of hemorrhage and necrosis should raise the suspicion of GBM.17
Histologic identification of the anaplastic astrocytoma often depends upon the presence of mitotic activity within a glial population that also demonstrates nuclear pleomorphism. The distinction is of significance in that most protocols recommend aggressive chemotherapeutic and radiation therapy to high-grade tumors (WHO grades III and IV) while some protocols suggest watchful waiting for low-grade neoplasms (WHO grades I and II). Ki-67 immunohistochemistry can be used to identify those tumors with proliferating cells, and, more importantly, in mitosis. Tumors that are excessively cellular with hyperchromatic, elongated nuclei may be designated anaplastic astrocytomas even in the absence of mitotic figures if the biopsy is small and associated with a brisk Ki-67 labeling index > 5.
The distinction from oligodendroglioma is important in that mitotic activity is not as reliable in identifying anaplastic change in oligodendroglial neoplasms. The cytoplasm of astrocytic neoplasms is probably the most reliable clue in distinguishing between these tumors, particularly in large specimens. The cytoplasm in astrocytoma is simplified from that seen in the reactive astrocyte with its abundant cytoplasm and complex branching processes, highlighted by GFAP. The neoplastic astrocytic cytoplasm can be either unipolar or bipolar, pulled away from the nucleus in one or two directions. The irregular tumor nuclei are embedded in an abundant eosinophilic fibrillar, or homogeneous, background. An exception to this rule is found in gemistocytic astrocytomas that reveal a plump cytoplasmic body with an eccentric nucleus. While most gemistocytic neoplasms will disclose frequent mitotic activity, there are documented cases lacking mitotic activity and prolonged survival, so grading of these variants is necessary.
The anaplastic astrocytoma is usually diffusely and strongly immunoreactive for the GFAP and S-100, revealing the simple elongated cell processes. Immunohistochemistry for Ki-67 has been shown to be prognostically useful in characterizing aggressive neoplasms.21 The labeling index of anaplastic astrocytic neoplasms is often three times greater than that seen in well-differentiated astrocytic neoplasms and approximately half that found in GBMs. At Duke University, diffuse astrocytomas (grade II) commonly exhibit a labeling index of less than 4% and usually around 1%, while grade III–IV astrocytomas (anaplastic astrocytomas and GBMs) exhibit a labeling index of greater than 5%. It is our practice to grade tumors based on histologic morphology and secondarily comment on the labeling index.
A comment should be offered regarding our approach to using radiographic input in grading. While it is clear that most tumors that enhance on computed tomography (CT) or magnetic resonance imaging (MRI) scanning are high-grade, some tumors that only exhibit increased T2 or fluid-attenuated inversion recovery (FLAIR) signal may hide an anaplastic astrocytoma. If one encounters a low-grade astrocytoma on a biopsy of a lesion that radiographically shows enhancement, one might want to use the non-committal term of infiltrating astrocytoma and comment on the adequacy (or lack thereof) of the biopsy.
Glioblastoma
The most common primary brain tumor16 encountered in the adult is GBM. Constituting from one-third to one-half of all brain tumors (and higher among those coming to biopsy), GBM is the most malignant form of glial neoplasia and may arise anywhere in the central nervous system. Inherited tendencies to GBM occur in Li–Fraumeni syndrome (TP53 gene)22 and familial colonic adenocarcinoma syndrome (deficiency of DNA mismatch repair enzymes).16,23
GBM is a highly invasive, rapidly growing glial neoplasm characterized grossly by hemorrhage, necrosis, and extensive infiltration (Figure 7.2a). Blurring of tumor/brain interfaces and gray–white junctions, lateral herniations, and obliteration of sulci reflect neoplastic permeation of tissues by tumor and associated edema.
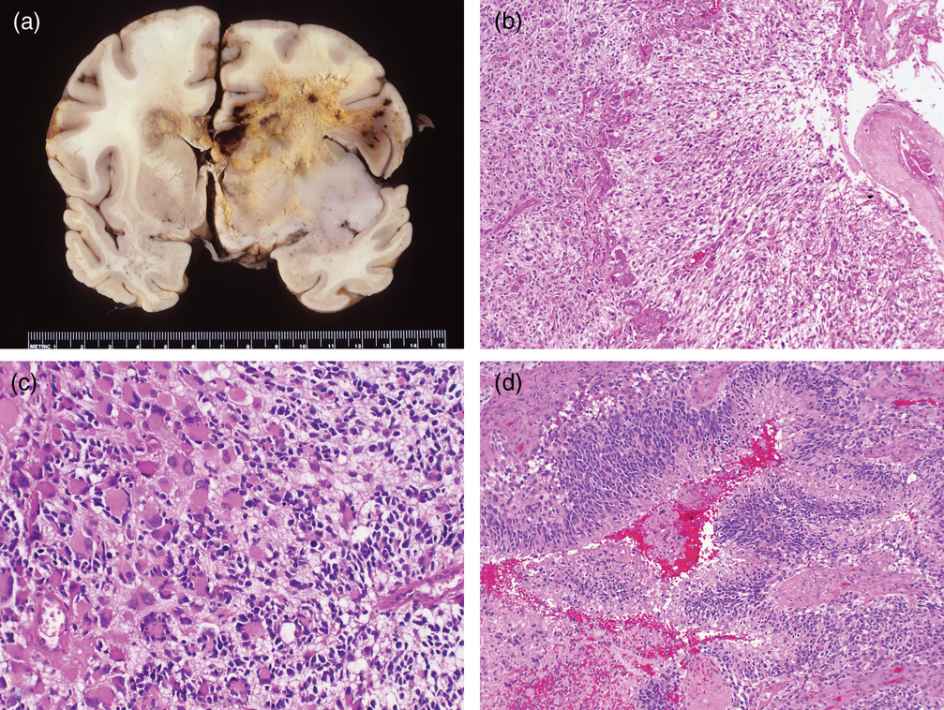
(a) Grossly, glioblastoma is characterized by bihemispheric spread and extensive hemosiderin staining and multifocal hemorrhages along with irregular necrosis.
(b) This glioblastoma exhibits elongated fibrillar cells oriented towards a necrotic focus and ringed by proliferating vessels.
(c) Cytologic heterogeneity is marked in this example, with small anaplastic cells infiltrating among the large, pleomorphic, multinucleated tumor cells
(d) This dense collection of irregular pleomorphic cells with very high nuclear to cytoplasmic ratios mimics focally the cytology of a primitive neuroectodermal tumor as the cellspseudopalisade about a focus of necrosis.
A spectrum of histologic forms ranging from small, tightly packed, elongated cells (Figure 7.2b) to huge, monstrocellular forms with bizarre nuclear profiles (Figure 7.2c) is encountered frequently among any group of GBMs.24 Even in the face of significant cytologic heterogeneity, the glial origin of the tumor cells is rarely questioned. Cytologically, as seen from touch and smear preparations, the tumor cells are loosely gathered and readily reveal fibrillar processes. In histologic preparations, GBM will frequently demonstrate a fibrillar background with minimal intercellular cohesion. Single-cell infiltration into the brain parenchyma is characteristic of glial neoplasia. Although a number of grading systems of glioma are extant, vascular proliferation and mitotic activity define histologic landmarks of high-grade astrocytoma in virtually all grading systems (Kernohan system excepted) with necrosis surrounded by pseudopalisading tumor cells verifying the high grade (Figure 7.2b and d).2 Despite writings to the contrary, mitotic figures may be scarce in even the most straightforward high-grade gliomas; here, the proliferative potential may be uncovered by immunoreactivity to the Ki-67 antigen as determined by the various monoclonal antibodies to the antigen, including Mib-1. While high-grade gliomas display proliferative indices threefold higher than low-grade gliomas, the labeling index with Mib-1 for a GBM is often as low as 5%.21
Diagnosing a GBM is not a challenge in the majority of cases. However, a certain, not insignificant, minority do cause diagnostic problems, particularly at the time of frozen section. Because the surgeon wants to perform a complete resection of a metastatic tumor but is often satisfied with less than a gross total resection of a glioma, the distinction has some importance.25
Glioblastomas in adults cause diagnostic problems when their histologic patterns mimic the following: sarcomas, meningiomas, carcinomas, lymphomas, pleomorphic xanthoastrocytomas (PXAs), and ependymomas.17,25 Even in those cases where the mimicry is faithful by light microscopy, GFAP staining usually belies histogenesis, revealing reactivity in at least some cells of greater than 90% of GBM. Another useful clue is found along the margin of the tumor, where the propensity of gliomas is to widespread infiltration into the surrounding brain by individual malignant cells, in contrast to metastatic tumors, where there is a sharp interface between malignant cells and a thin border of benign, inflamed reactive brain tissue. Rare GBMs will exhibit a pseudopapillary arrangement due to perivascular preservation in necrosis that is reminiscent of an adenocarcinoma, but GFAP reactivity is diagnostic. Care must be taken to use only the low-molecular-weight keratin, CAM5.2, because higher-weight keratins can be found in all astrocytomas.
Occasionally a GBM will be superficial and parasitize the overlying meningeal vascular supply. Epithelial membrane antigen is often positive in meningiomas (and negative in GBM), but may highlight reactive meningothelial cells along the attachment regions.
Lymphoma mimic
A pseudolymphoma pattern may result from glioma cells invading the perivascular Virchow–Robin space. In this way, tumor cells come to lie in the perivascular spaces of virgin brain to be found by a needle that has strayed beyond the confines of the tumor. This perivascular pattern may also be seen in the lobe contralateral to a large primary brain tumor in which clinicians are trying to rule out distant spread versus radiation effect. Immunohistochemistry for GFAP, CD45, and CD20 can easily resolve this dilemma with gliomas being GFAP-reactive and CD45/CD20-negative and lymphomas vice versa.
Pleomorphic low-grade glioma mimic
The most significant problem in evaluating glial tumors is in the distinction from WHO grade I–II tumors. The three most common mimics of GBM are pilocytic astrocytoma, PXA, and desmoplastic infantile ganglioglioma (DIG); the issue is significant because survival without radiation therapy is usually prolonged in all three of the low-grade tumors, making toxicity from radiation therapy an issue. On MRI, all three tumors enhance remarkably with gadolinium, revealing sharply circumscribed borders with the brain. Each of these tumors is described below in their respective sections.
Oligodendroglioma
Well-differentiated oligodendroglioma
Oligodendrogliomas constitute anywhere from 5% to 10% of all intracranial gliomas according to published results.16,17 Although oligodendrogliomas are unusually diagnosed in childhood, it is common to identify them in young adults who have a longstanding history of seizures dating back to childhood and, more likely than not, related to the onset of the tumor in childhood. Most tumors are identified in the fourth and fifth decades. No inherited or familial tendency to oligodendrogliomatosis is known.
The cerebral hemisphere accounts for the great majority of oligodendrogliomas; however, they may also be found in other regions. The true incidence of oligodendrogliomas in cerebellum, brainstem, and sites such as the ventricles and optic nerves is undoubtedly overstated because of the presence of cells in juvenile pilocytic astrocytomas, glial-neurocytic tumors, and diffuse astrocytomas that are microscopically similar to oligodendroglial cells.
Oligodendroglioma most commonly arises in the frontal or parietal lobes.26 The cut surface of the tumor may be well-defined and reveals mucinous change resulting in a gelatinous appearance to the tumor. Gritty microcalcifications may be encountered, although necrosis and cystic degeneration are seldom conspicuous.17
Microscopically, the artifactual, “fried egg” perinuclear cytoplasmic haloes (Figure 7.3a and b) are not present in small samples that are rapidly fixed or are submitted to frozen section prior to formalin fixation. While this degenerative process involves the cytoplasm, the nucleus is not readily affected. Rather, the nucleus exhibits a thin delicate chromatin pattern with punctate condensations of chromatin (“chromocenters”) and usually no evidence of an eosinophilic nucleolus. Mitotic activity may be surprisingly frequent in tumors that are only moderately cellular. Occasionally, otherwise well-differentiated oligodendrogliomas will exhibit preservation of the cytoplasm and an eosinophilic droplet of cytoplasm forming so-called mini-gemistocytes; this has no prognostic importance. Occasionally, an entire section of the tumor may be composed of these mini-gemistocytes and result in confusion with the astrocytoma.
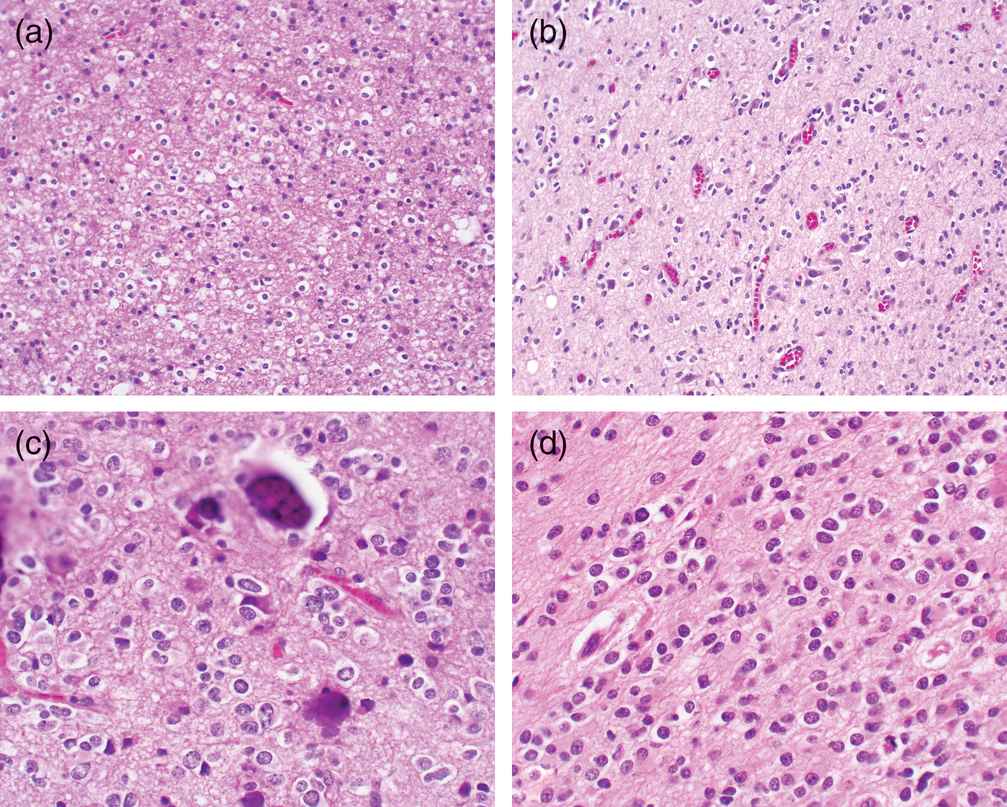
(a) Histologically, well-differentiated oligodendroglioma cells are recognized by their perinuclear haloes forming the “fried egg” appearance.
(b) Here oligodendroglioma cells satellite about vessels.
(c) Microcalcifications are commonly encountered in oligodendrogliomas.
(d) Treatment can render the oligodendroglioma “astrocytoma-like” with loss of haloes and pleomorphic nuclei.
Another feature of oligodendrogliomas is pools of mucin, presumably a degenerative phenomenon. When present, the thin, delicate, arcuating vascular pattern is reassuring for the diagnosis of oligodendroglioma. However, we depend on nuclear detail primarily to distinguish oligodendroglial tumors from astrocytic tumors. The nuclei of oligodendrogliomas are round to oval and occasionally exhibit rounded nuclear lobulations. Within the nuclear envelope, the chromatin is distinctly thread-like with punctate hematoxylinophilic chromocenters evident; only rarely are eosinophilic nucleoli seen. Supportive evidence of oligodendroglial origin is the presence of perinuclear haloes as well as the formation of the secondary structures originally described by Scherer,27 in which the oligodendroglial cells satellite about neurons and vascular structures (Figure 7.3c). Oligodendrogliomas are prone to subarachnoid accumulations, resulting in an unusual appearance of hypercellular white matter, modestly hypercellular cortical tissue, and densely hypercellular subarachnoid tumor. Tumors with these features usually, but not always, are also associated with arcuating thin delicate vessels and punctate cortical microcalcifications (Figure 7.3c).
Immunohistochemistry
Immunoreactivity for GFAP may be found in classic oligodendrogliomas and is especially pronounced in the mini-gemistocytes. Although a number of other antibodies have been put forth as exhibiting oligodendroglial reactivity, including Nogo-A and alpha-internexin, there has been no consistent antigenic marker for sensitively and specifically identifying tumors of oligodendroglial origin. Although of unclear origin and source, most antibodies to synaptophysin apparently cross-react with elements in oligodendrogliomas, resulting in a faint membrane-related reactivity, with rare examples exhibiting pronounced reactivity.
A number of biological problems have yet to be solved with regard to clearly identifying specific and sensitive markers for oligodendroglial tumors. A striking finding among molecular biological studies on gliomas is the extremely high incidence of loss of heterozygosity on 1p and 19q among oligodendrogliomas. Among the tumors with 19q loss is the concomitant mutation of CIC while FUBP1 mutations are found in some tumors with 1p loss. A very tight correlation exists between 1p,19q allelic losses and mutations of IDH1/2 as well as with mutations in the promoter region of TERT.28,29 In certain studies, these findings are present in up to 85% of oligodendroglial tumors and correlate with prolonged survivals, even among histologically similar oligodendroglial tumors. Further studies reviewing the incidence of these markers in therapeutic trial of gliomas are needed before the utility of these markers can be fully appreciated.
Anaplastic oligodendroglioma
The grading of oligodendrogliomas is a controversial subject and intimately intertwined with the issues of mixed oligoastrocytomas and the significance of grading the astrocytic component. Nonetheless, it is clear that certain tumors of pure oligodendroglial origin become markedly hypercellular, exhibit frequent mitoses, and are associated with necrosis with or without pseudopalisading of tumor cells. The vessels within these tumors similarly are more exuberant and may exhibit glomeruloid formations. It is important to note that some otherwise typical oligodendrogliomas may exhibit densely cellular nodules with brisk Ki-67 labeling; these tumors may be accepted as low-grade gliomas.
The recent revision of the WHO grading system recognizes the presence of a grade III anaplastic oligodendroglioma with the features described. At present, studies suggest that oligodendroglial tumors exhibiting nuclear atypia, significant mitotic activity, vascular proliferation, vascular hypertrophy, and necrosis should be classified as anaplastic oligodendrogliomas.30 The recent addition of alkylating chemotherapeutics such as temozolomide has apparently prolonged survival in patients with oligodendroglial tumors exhibiting 1p/19q losses, with median survivals of patients with either grade II or grade III tumors exceeding 9 years in some studies.31 Biopsies of these recurrent, treated oligodendrogliomas may be mistaken for astrocytomas due to the loss of perinuclear haloes and pleomorphism of the nuclei (Figure 7.3d). Reassuringly, these tumor continue to exhibit the 1p,19q co-deletions.
Mixed oligodendroglial and astrocytic neoplasms
Clinical studies justify the effort involved in distinguishing oligodendroglial tumors from astrocytic neoplasms. In particular, these studies have indicated that oligodendroglial tumors are more responsive to alkylator or nitrosourea chemotherapeutic drugs, drugs which are less effective among astrocytic neoplasms. Furthermore, other studies suggest that both oligodendroglial and astrocytic neoplasms exhibit identical mutations affecting IDH1/2, an observation that suggests a common progenitor cell among these tumors. The findings suggest mixed oligoastrocytic neoplasms may exist, a finding with mixed support from molecular data.
In the discussion of glial neoplasms, much emphasis is placed on nuclear characteristics in distinguishing astrocytic neoplasms from oligodendroglial. This distinction is fraught with controversy and is reflected in numerous studies which indicate a greater than 5% variation amongst neuropathologists in distinguishing between these two histologic subtypes.32
The WHO classification scheme recognizes the existence of gliomas with mixed morphologies and reflects the renewed interest arising in identifying uniform criteria for their diagnosis.33 The WHO defines the oligoastrocytoma as a “tumor with a conspicuous mixture of neoplastic oligodendrocytes and astrocytes, either diffusely intermingled or separated into distinct areas.” These authorities further warn, “tumors containing cells with oligodendroglial nuclei and small eccentric GFAP-positive cell bodies are not considered to be mixed gliomas.”33 While the authors tacitly accept the concept that oligodendrogliomas have nuclear features that are distinctive from astrocytes, they decline any further elaboration.
The Duke team led by Hai Yan has made significant discoveries that shed light on this controversy. The team has found that oligodendroglial tumors exhibit mutations in both IDH1/2 and TERT, findings which have been applied to tumors called oligoastrocytomas. Tumors with the oligodendroglial genotype with both IDH1/2 and TERT mutated exhibited significantly prolonged survivals versus those with either IDH1/2 or TERT alone. Further investigations are needed to determine the correlation of these genetic findings with the cytogenetic findings of single losses of either 1p or 19q in tumors morphologically called mixed oligoastrocytomas. Recent publications by a German group suggest that a clue may lie in CIC mutations found on mixed gliomas with 19q losses, such that those with CIC mutations survived longer than those with wild-type CIC. The Yan data suggest that most mixed gliomas will exhibit a divergent mutation pattern with the oligodendroglia genotype being present uncommonly.28,29
Grading mixed gliomas is controversial for these reasons. While it is clear that the Daumas-Duport guidelines of nuclear pleomorphism, mitotic activity, vascular proliferation, and necrosis are useful in grading most gliomas, it is not clear whether each population should be graded individually or together.34,35 Before grading, is it more important to know the 1p,19q status or the IDH1/2 and TERT status? How much heterogeneity is present in these mixed gliomas and how many blocks need to be tested? These questions are the focus of several laboratory groups and should be answered soon.
Circumscribed astrocytomas
Pilocytic astrocytoma
Pilocytic astrocytomas (WHO grade I) represent the most common glioma found in young adults,16,36 and are among the most common solid tumors in these age groups. The tumor apparently favors neither sex nor race in its incidence. The tumors seem to prefer the midline and the periventricular regions, with occasional examples producing lobular intraventricular masses.17 Optic nerve tumors are most commonly pilocytic astrocytomas. Pilocytic astrocytomas arising in the brainstem tend to be exophytic masses and may arise in either the medulla or the midbrain.
In children, cerebellar, diencephalic, and optic nerve low-grade astrocytomas are predominantly of pilocytic histogenesis.37 Furthermore, the majority of spinal cord tumors in children are pilocytic. Macroscopically, the pilocytic astrocytoma exhibits a soft, opalescent appearance in a discrete mass that the surgeon describes as “peeling away” from the surrounding parenchyma. Longstanding cases may exhibit calcium or are speckled with rusty hemosiderin. In the cerebellum, the tumor is commonly associated with a cyst that can be quite large, overwhelming the small mural nodule that represents the neoplasm. In the optic nerve, though the tumor histologically exhibits a more infiltrative quality, the gross appearance is that of a well-defined mass within the optic nerve.
Microscopically, Verhoeff38 described three patterns of pilocytic astrocytoma. Although his descriptions were based originally on optic nerve gliomas, these three patterns are also seen within pilocytic astrocytomas arising in other sites. The three patterns are “coarsely reticulated with microcystic foci,” or the so-called biphasic pattern; “finely reticulated”; and a third controversial pattern, termed “adult.” Typical tumors exhibit multiple patterns. The most common pattern outside of the optic nerve corresponds to Verhoeff’s “coarsely reticulated with microcystic foci,” or the so-called biphasic morphology (Figure 7.4a). This pattern is composed of spindle, often bipolar, astrocytes that form bundles and are coarsely woven together. The tumor cells collar around prominent blood vessels and may approach the appearance of the ependymal pseudorosette. The free cytoplasmic ends of the cells do not span the distance between blood vessels, leaving degenerate microcystic foci. The blood vessels are usually prominent and appear hypertrophic but may be densely hyalinized. In younger patients (less than 18 years of age), mitotic figures may be found but are usually not numerous.17 Instead, Rosenthal fibers, eosinophilic granular bodies, and protein droplets may be quite conspicuous. In some examples, a pilocytic component may be the glial aspect of a ganglioglioma. Although perivascular lymphocytes may be encountered in pilocytic astrocytomas, their common occurrence in gangliogliomas should prompt a search for dysmorphic ganglion cells. Some examples of pilocytic astrocytomas, particularly those arising in the hypothalamus, will exhibit radial perivascular orientation, reminiscent of the ependymoma. Many such examples will demonstrate abundant intercellular mucin. These examples will typically lack Rosenthal fibers and eosinophilic granular bodies and are known as pilomyxoid astrocytomas (Figure 7.4b), a tumor said to be more clinically aggressive and deserving of the WHO grade II designation.33 The pilocytic astrocytoma tends to have diffuse GFAP reactivity, in contrast to the ependymoma, in which pronounced GFAP is limited to the perivascular processes. Furthermore, epithelial membrane antigen reactivity in small perinuclear vesicles should raise the possibility of ependymoma.
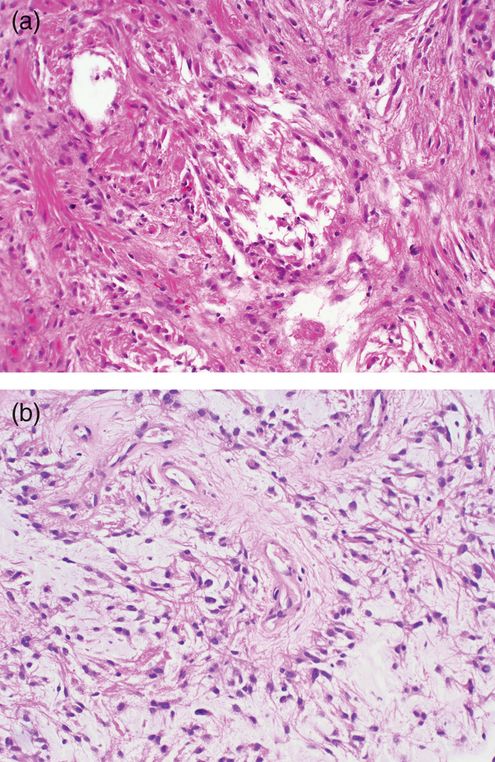
(a) Pilocytic astrocytomas can exhibit a biphasic appearance of densely packed cells separated by loosely cellular regions with microcystic formation.
(b) The pilomyxoid variant features radially oriented tumor cells and a lack of Rosenthal fibers.
Verhoeff’s38 second pattern, most commonly found within the optic nerve but also found in the cerebellum and hypothalamus, is the “finely reticulated” variant. Within the optic nerve, this pattern suggests an exaggeration of the normal neuroglial pattern. Rosenthal fibers and eosinophilic granular bodies are not as commonly encountered. Nevertheless, the neoplastic nuclei are monotonous and bland, and lack mitotic figures, readily bringing to mind a low-grade process.39 It is this variant of pilocytic astrocytoma with which benign reactive gliosis is most commonly confused. Indeed, the pathologist is occasionally at the mercy of the neurosurgeon and neuroradiologist in evaluating these lesions because of the close mimicry between pilocytic astrocytomas and reactive gliosis in the hypothalamic and pineal regions.
The third pattern, which may be termed “adult,” can be mistaken for high-grade fibrillary astrocytoma because of the mildly pleomorphic, hyperchromatic nuclei with coarse chromatin in which rare mitotic figures can be encountered. The tumor is largely solid and hyalinized tortuous blood vessels complete the picture.40,41 Necrosis may also be encountered and is usually of the coagulative type, strongly suggestive of an ischemic origin. In this pattern, the brightly eosinophilic Rosenthal fibers, protein droplets, and eosinophilic granular bodies assist the wary, as does its radiographic image of a well-circumscribed, brightly enhancing mass.
Within any one pilocytic astrocytoma, multiple microscopic patterns may be encountered. However, the biphasic pattern referred to by most neuropathologists represents the alternating solid and microcystic pattern seen in Verhoeff’s first pattern. Occasionally, in small biopsy samples, it may be impossible to distinguish the pilocytic astrocytoma from a diffusely infiltrating fibrillary astrocytoma or from an ependymoma. While epithelial membrane antigen may be helpful in the latter, the former may require radiology that may reveal features of strong, diffuse enhancement and relatively sharp circumscription of the pilocytic astrocytoma. One may be left recommending either “watchful waiting” or a more generous sample.
Of particular importance in recognizing most pilocytic astrocytomas are the four features that characteristically participate in variable amounts: microcyst formation, eosinophilic granular bodies, Rosenthal fibers, and protein droplets. Microcystic change is a degenerative feature common to all forms of glioma but can be prominent, relegating the tumor to a mural nodule within the cyst. Adjacent to these cysts, the cells assume a clear-cell, “oligodendroglial” appearance. Membrane-bound eosinophilic granules appear to correspond to numerous small autophagic vacuoles within the tumor astrocytes. Rosenthal fibers are elongated, brightly eosinophilic, hyalinized, corkscrew structures that are composed of α-B-crystalline.42
Recent molecular studies indicate that most pilocytic astrocytomas, particularly of the posterior fossa, exhibit a duplication, truncation of BRAF with the KIAA gene, both of which are located on chromosome 7q, resulting in constitutive activation of the BRAF gene. In neurofibromatosis type 2, BRAF is constitutively activated by a germline mutation in the NF2 gene, which is biologically responsible for BRAF deactivation.43
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


